Advanced Synthesis of Fluorescent Dehydroabietyl Thiazolinone Derivatives for Pharmaceutical Applications
The pharmaceutical and fine chemical industries are constantly seeking novel scaffolds that combine structural complexity with functional utility, particularly in the realm of fluorescent probes and anticancer agents. Patent CN110878068B introduces a groundbreaking synthetic methodology for producing Dehydroabietyl-B Cyclothiazole-Imino-(Benzylidene) Thiazolinone, a sophisticated heterocyclic compound derived from natural rosin resources. This innovation represents a significant leap forward in valorizing forest chemical byproducts, transforming simple diterpene resin acids into high-value bioactive intermediates. The core breakthrough lies in the strategic fusion of a thiazole ring with the rigid dehydroabietic acid skeleton, creating an extended conjugated system that endows the molecule with unique fluorescence properties. 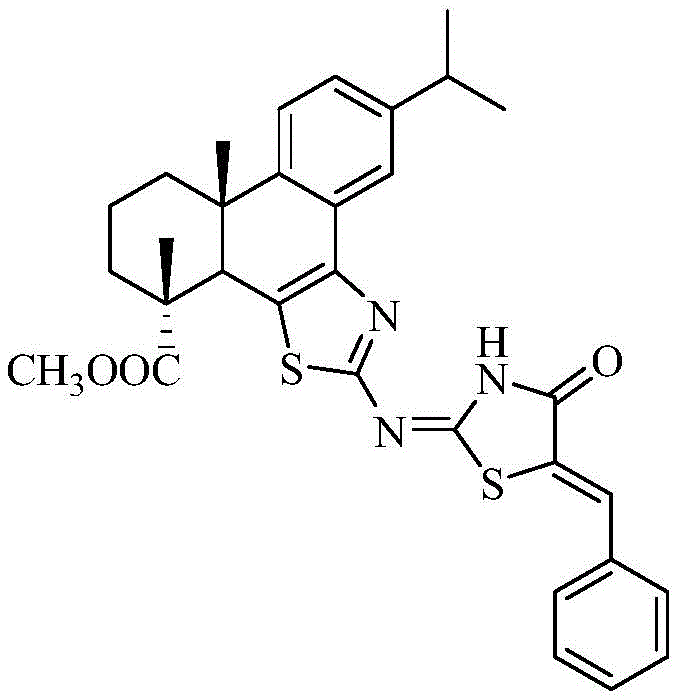 For R&D directors and procurement specialists, this patent offers a compelling case study in sustainable chemistry, demonstrating how abundant natural feedstocks can be engineered into precision tools for drug discovery and medical diagnostics without compromising on performance or scalability.
For R&D directors and procurement specialists, this patent offers a compelling case study in sustainable chemistry, demonstrating how abundant natural feedstocks can be engineered into precision tools for drug discovery and medical diagnostics without compromising on performance or scalability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of complex thiazole-fused heterocycles often relies on multi-step sequences involving expensive transition metal catalysts or harsh reaction conditions that generate significant hazardous waste. Conventional routes to similar fluorescent scaffolds frequently utilize petrochemical-derived aromatics, which are subject to volatile market pricing and supply chain instability. Furthermore, introducing specific functional groups onto rigid polycyclic backbones typically requires protecting group strategies that add unnecessary steps, reduce overall atom economy, and complicate purification processes. In the context of rosin derivatives, previous modification attempts have often struggled to achieve high regioselectivity or have resulted in products with limited solubility and bioavailability. These legacy methods create bottlenecks for reliable pharmaceutical intermediate supplier networks, as the cost of goods sold remains prohibitively high for widespread screening applications. The reliance on non-renewable feedstocks also conflicts with the growing industry mandate for green chemistry and sustainable manufacturing practices.
The Novel Approach
The methodology disclosed in CN110878068B circumvents these historical challenges through a streamlined, four-step linear synthesis that maximizes efficiency and minimizes environmental impact. By starting with methyl 7-carbonyl dehydroabietate, a readily available derivative of dehydroabietic acid, the process leverages the inherent chirality and rigidity of the natural product to direct subsequent transformations. The key innovation involves a tandem bromination-cyclization sequence that efficiently constructs the benzothiazole moiety directly onto the B-ring of the diterpene skeleton. 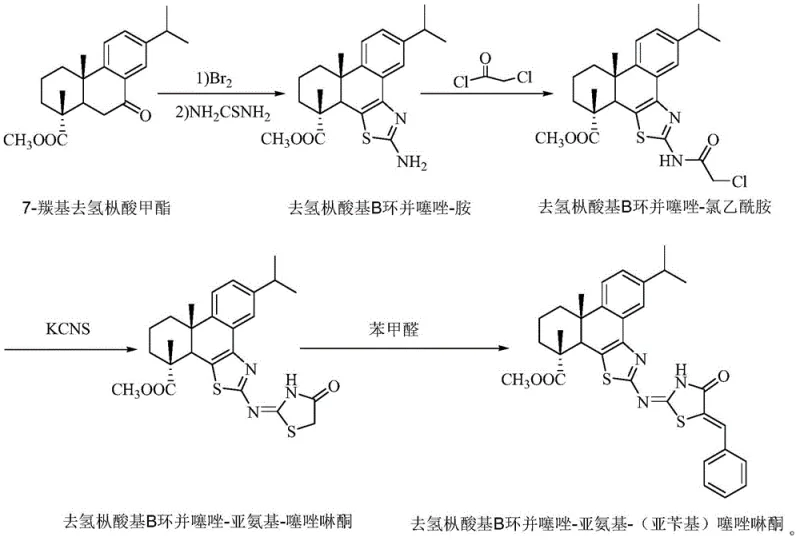 This approach eliminates the need for pre-functionalized aromatic building blocks, significantly reducing cost reduction in fine chemical manufacturing. The subsequent installation of the thiazolinone ring via chloroacetylation and thiocyanate cyclization proceeds under mild conditions, preserving the integrity of the sensitive ester functionality. Finally, the condensation with benzaldehyde extends the conjugation, locking in the fluorescent properties. This logical, step-wise progression ensures high purity and reproducibility, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
This approach eliminates the need for pre-functionalized aromatic building blocks, significantly reducing cost reduction in fine chemical manufacturing. The subsequent installation of the thiazolinone ring via chloroacetylation and thiocyanate cyclization proceeds under mild conditions, preserving the integrity of the sensitive ester functionality. Finally, the condensation with benzaldehyde extends the conjugation, locking in the fluorescent properties. This logical, step-wise progression ensures high purity and reproducibility, making it an ideal candidate for commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Thiazole-Thiazolinone Fusion Chemistry
The mechanistic pathway begins with the electrophilic bromination of the ketone alpha-position in methyl 7-carbonyl dehydroabietate, activated by the acidic medium of glacial acetic acid. This alpha-bromo ketone serves as a highly reactive electrophile, poised for nucleophilic attack by thiourea. Upon heating to 105°C, the sulfur atom of thiourea attacks the carbonyl carbon while the nitrogen attacks the alpha-carbon, facilitating a dehydration-cyclization cascade that aromatizes the newly formed thiazole ring. This step is critical as it establishes the planar conjugated system necessary for fluorescence. The resulting amine intermediate is then acylated with chloroacetyl chloride in the presence of triethylamine, which acts as a proton scavenger to drive the formation of the amide bond. The chlorine atom on the acetamide side chain remains intact, serving as a leaving group for the subsequent intramolecular cyclization. When treated with potassium thiocyanate in refluxing ethanol, the thiocyanate ion displaces the chloride, and the resulting intermediate undergoes rapid cyclization to form the 2-imino-4-thiazolidinone ring. This dual-heterocycle architecture creates a rigid, electron-rich environment that stabilizes the excited state of the molecule.
Impurity control is meticulously managed through specific solvent systems and purification protocols defined in the patent. For instance, the initial benzothiazole-amine is purified using a dichloromethane-methanol gradient, targeting a specific TLC Rf value between 0.23 and 0.34 to ensure the removal of unreacted starting materials and poly-brominated byproducts. Similarly, the final condensation step with benzaldehyde is monitored to prevent the formation of bis-condensation products or unreacted aldehyde adducts. The use of silica gel column chromatography with precise ethyl acetate-petroleum ether ratios allows for the isolation of the target compound with high homogeneity. This rigorous attention to purification parameters is essential for maintaining the stringent purity specifications required for biological testing and potential therapeutic applications. The mechanism ensures that the chiral centers at C4 and C10 of the dehydroabietic acid backbone remain unaffected, preserving the stereochemical integrity of the natural product scaffold throughout the synthesis.
How to Synthesize Dehydroabietyl Thiazolinone Efficiently
Executing this synthesis requires careful attention to reaction stoichiometry and temperature control to maximize yield and minimize side reactions. The process is designed to be robust, utilizing common laboratory reagents and standard glassware, which simplifies the technology transfer from R&D to pilot plant operations. The initial bromination step must be controlled to prevent over-bromination, while the cyclization with thiourea requires sustained heating to ensure complete ring closure. Subsequent acylation and thiocyanate displacement steps are exothermic and benefit from dropwise addition of reagents to manage heat evolution. The final condensation with benzaldehyde drives the equilibrium towards the product through the removal of water, often facilitated by the reflux conditions. Detailed standardized synthesis steps see the guide below for precise operational parameters.
- Bromination and Cyclization: React methyl 7-carbonyl dehydroabietate with bromine in glacial acetic acid, followed by thiourea addition at 105°C to form the benzothiazole-amine intermediate.
- Acylation: Treat the amine intermediate with chloroacetyl chloride and triethylamine in dichloromethane at room temperature to yield the chloroacetamide derivative.
- Thiazolinone Ring Formation: Reflux the chloroacetamide with potassium thiocyanate in ethanol to close the second heterocyclic ring, forming the imino-thiazolinone core.
- Condensation: React the imino-thiazolinone with benzaldehyde in ethanol under reflux conditions to finalize the conjugated system and obtain the target fluorescent compound.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this synthetic route offers distinct advantages rooted in the abundance and renewability of its primary feedstock. Dehydroabietic acid is a major component of disproportionated rosin, a commodity chemical produced in vast quantities globally, ensuring a stable and secure supply chain that is less susceptible to the geopolitical volatility affecting petrochemical derivatives. The elimination of precious metal catalysts such as palladium or platinum from the synthesis not only reduces the direct material cost but also removes the complex and expensive downstream processing steps associated with heavy metal removal. This simplification translates directly into substantial cost savings and a faster time-to-market for the final active ingredient. Furthermore, the use of common organic solvents like dichloromethane, ethanol, and ethyl acetate aligns with existing solvent recovery infrastructure in most chemical manufacturing facilities, enhancing operational efficiency.
- Cost Reduction in Manufacturing: The process achieves significant economic efficiency by utilizing low-cost natural resin acids as the starting framework, effectively bypassing the need for expensive synthetic aromatic precursors. By avoiding transition metal catalysis, the method eliminates the capital expenditure associated with catalyst recovery systems and the operational costs of meeting strict residual metal limits in pharmaceutical products. The linear nature of the synthesis minimizes the number of isolation and purification stages, which reduces solvent consumption and labor hours per kilogram of product. Additionally, the high atom economy of the cyclization steps ensures that a greater proportion of the raw material mass is incorporated into the final product, further driving down the cost of goods.
- Enhanced Supply Chain Reliability: Sourcing raw materials from the forestry sector provides a diversified supply base that complements traditional petrochemical supply chains, mitigating risks associated with oil price fluctuations. The intermediates generated in the early stages of the synthesis, such as the benzothiazole-amine, are stable and can potentially be stockpiled, allowing for flexible production scheduling and rapid response to demand spikes. The robustness of the reaction conditions, which do not require cryogenic temperatures or high-pressure equipment, means that production can be easily distributed across multiple manufacturing sites with standard capabilities. This decentralization capability strengthens the overall resilience of the supply network against localized disruptions.
- Scalability and Environmental Compliance: The synthetic protocol is inherently scalable, relying on unit operations like reflux and distillation that are well-understood and easily modeled for larger reactor volumes. The absence of toxic heavy metals simplifies waste stream management, reducing the environmental burden and compliance costs associated with hazardous waste disposal. The process generates primarily organic waste that can often be incinerated for energy recovery or treated via standard biological wastewater treatment methods. This alignment with green chemistry principles not only satisfies regulatory requirements but also enhances the corporate sustainability profile of manufacturers adopting this technology, appealing to environmentally conscious stakeholders.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this novel fluorescent compound. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear understanding of the technology's value proposition. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this intermediate into their own development pipelines.
Q: What is the primary raw material source for this fluorescent compound?
A: The synthesis utilizes dehydroabietic acid, a natural component of rosin (pine resin), making it a renewable and cost-effective starting material compared to fully synthetic petrochemical precursors.
Q: Does the final compound exhibit specific biological activity?
A: Yes, testing indicates the compound possesses both fluorescence properties suitable for molecular recognition and significant anticancer activity against human tongue squamous cell carcinoma lines (cal27 and scc9).
Q: Is the synthetic route suitable for large-scale manufacturing?
A: The process relies on standard organic unit operations such as reflux, distillation, and silica gel chromatography, avoiding exotic catalysts, which facilitates straightforward scale-up from laboratory to commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dehydroabietyl Thiazolinone Supplier
As the demand for specialized fluorescent probes and anticancer intermediates grows, partnering with an experienced CDMO becomes essential for navigating the complexities of process development and scale-up. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from the benchtop to the marketplace. Our team is equipped with rigorous QC labs and advanced analytical instrumentation to meet stringent purity specifications, guaranteeing that every batch of dehydroabietyl thiazolinone derivative performs consistently in your biological assays. We understand the critical nature of supply continuity and are committed to delivering high-quality intermediates that accelerate your drug discovery timelines.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be optimized for your specific needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this rosin-based scaffold. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and expert evaluation. Together, we can unlock the full potential of natural product derivatives in the next generation of therapeutic and diagnostic agents.