Scalable Synthesis of Beta-L-FD4C Antiviral Intermediate via Optimized L-Xylose Route
Scalable Synthesis of Beta-L-FD4C Antiviral Intermediate via Optimized L-Xylose Route
The global demand for effective antiviral therapeutics targeting HIV and HBV continues to drive innovation in nucleoside analog manufacturing. Patent CN1329407C presents a groundbreaking methodology for the synthesis of beta-L-5-fluoro-2',3'-dideoxy-2',3'-didehydrocytidine, commonly known as beta-L-FD4C. This compound has been identified as a particularly promising agent for treating viral infections, yet historical synthetic routes have often suffered from low yields and complex purification challenges that hindered large-scale adoption. The disclosed invention addresses these critical bottlenecks by introducing a streamlined, seven-step sequence starting from the abundant and cost-effective chiral pool material, L-xylose. By leveraging solid acid catalysts and telescoped reaction conditions, this process not only enhances overall chemical efficiency but also aligns with modern green chemistry principles, making it an ideal candidate for industrial scale-up.
For procurement specialists and supply chain directors, the strategic value of this patent lies in its robust design for commercial viability. Unlike earlier academic protocols that relied on stoichiometric amounts of hazardous reagents or difficult-to-remove liquid acids, this method employs macroporous resin catalysts such as Amberlyst 15. This shift allows for simple filtration-based removal of the catalyst, thereby eliminating the need for extensive aqueous workups that generate significant wastewater volumes. Furthermore, the process minimizes the number of isolation steps; several intermediates are carried forward as solutions without evaporation to dryness. This operational fluidity translates directly into reduced cycle times and lower energy consumption, positioning this technology as a superior choice for reliable pharmaceutical intermediates supplier networks aiming to optimize their manufacturing footprint while ensuring consistent quality.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of beta-L-FD4C and related L-nucleoside analogs faced significant hurdles regarding yield and environmental impact. Early synthetic strategies, such as those described in foundational literature by Lin et al., often resulted in suboptimal conversion rates that made commercial production economically unfeasible. These conventional pathways frequently utilized harsh liquid mineral acids for protection and deprotection steps, which necessitated neutralization and generated large quantities of salt waste during workup. Additionally, the reliance on multiple isolation and drying steps between each transformation increased the risk of product degradation and introduced opportunities for impurity ingress. The cumulative effect of these inefficiencies was a high cost of goods sold (COGS) and a supply chain vulnerable to delays, rendering the final antiviral API prohibitively expensive for widespread therapeutic use in developing regions where HIV and HBV burdens are highest.
The Novel Approach
The methodology outlined in CN1329407C represents a paradigm shift towards process intensification and waste minimization. A key innovation is the use of cation exchange resins for acid-catalyzed reactions, which provides a heterogeneous catalytic system that is easily separable from the reaction mixture. This eliminates the corrosive handling issues associated with sulfuric or hydrochloric acid and drastically reduces the generation of inorganic salts. Moreover, the process is engineered to allow for solvent swaps and telescoping; for instance, the bisacetal intermediate can be hydrolyzed directly in the same vessel after a simple solvent adjustment, bypassing the need for complete isolation. This approach not only accelerates the production timeline but also preserves the integrity of sensitive intermediates that might degrade upon prolonged exposure to heat during drying. The result is a high-purity product stream that requires minimal downstream purification, offering substantial cost savings in antiviral manufacturing.
Mechanistic Insights into Resin-Catalyzed Acetal Formation and Coupling
The chemical elegance of this synthesis begins with the stereoselective protection of L-xylose. The reaction with acetone in the presence of a dehydrating agent like copper sulfate and an acid resin catalyst facilitates the formation of the 1,2-O-isopropylidene derivative. The mechanism involves the protonation of the carbonyl oxygen of acetone by the sulfonic acid groups on the resin, increasing its electrophilicity towards the diol moieties of the sugar. The macroreticular structure of the resin ensures that the active sites are accessible while preventing the entrapment of the product, which aids in high recovery rates. Following this, selective hydrolysis of the 5,6-acetal is achieved under mild acidic conditions, preserving the 1,2-protection while exposing the primary alcohol for subsequent functionalization. This precise control over protecting group orthogonality is crucial for maintaining the stereochemical integrity of the L-sugar backbone throughout the synthesis.
A critical juncture in the pathway is the glycosylation step, where the activated enose sugar couples with the protected nucleobase. 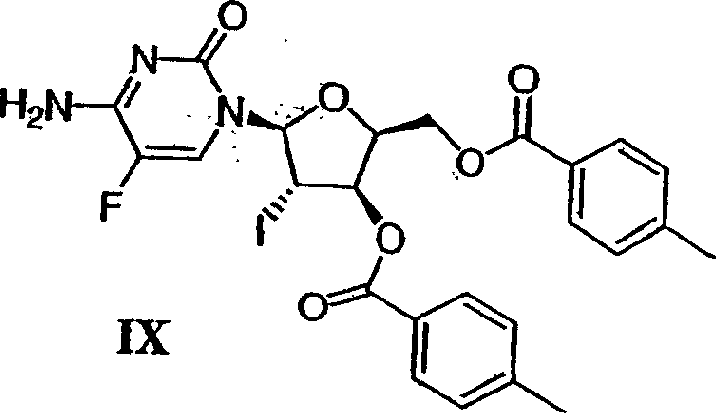 As illustrated in the reaction scheme, the enose intermediate is generated via an iodination-elimination sequence using triphenylphosphine and imidazole. This creates a reactive vinyl species that undergoes coupling with bis-trimethylsilyl-protected 5-fluorocytosine in the presence of N-iodosuccinimide (NIS). The NIS acts as a halogenating agent to activate the anomeric center, promoting the formation of the N-glycosidic bond with high beta-selectivity. The use of silyl protection on the cytosine ring prevents side reactions at the exocyclic amine and carbonyl oxygen, ensuring that the coupling occurs exclusively at the N1 position. Following coupling, reductive dehalogenation with zinc metal and acetic acid removes the iodine substituent and the toluoyl protecting groups simultaneously, streamlining the route to the final unsaturated nucleoside scaffold.
As illustrated in the reaction scheme, the enose intermediate is generated via an iodination-elimination sequence using triphenylphosphine and imidazole. This creates a reactive vinyl species that undergoes coupling with bis-trimethylsilyl-protected 5-fluorocytosine in the presence of N-iodosuccinimide (NIS). The NIS acts as a halogenating agent to activate the anomeric center, promoting the formation of the N-glycosidic bond with high beta-selectivity. The use of silyl protection on the cytosine ring prevents side reactions at the exocyclic amine and carbonyl oxygen, ensuring that the coupling occurs exclusively at the N1 position. Following coupling, reductive dehalogenation with zinc metal and acetic acid removes the iodine substituent and the toluoyl protecting groups simultaneously, streamlining the route to the final unsaturated nucleoside scaffold.
How to Synthesize Beta-L-FD4C Efficiently
Executing this synthesis requires careful attention to reaction conditions, particularly temperature control during the exothermic acylation and coupling steps. The protocol dictates the use of specific solvent systems, such as dichloromethane or toluene mixtures, to maintain solubility of intermediates while facilitating easy removal of byproducts. For R&D teams looking to implement this technology, the key lies in optimizing the telescoping sequences to minimize solvent usage. The detailed standard operating procedures for each transformation, including precise molar ratios of reagents like zinc dust and ammonium sulfate, are critical for reproducing the high yields reported in the patent examples. The following guide outlines the standardized synthesis steps derived from the patent data to ensure reproducibility and safety in a pilot or commercial plant setting.
- Protect L-xylose with acetone using an acid catalyst like Amberlyst 15 to form the bisacetal, followed by selective hydrolysis.
- Acylate the alcohol moiety with p-toluoyl chloride, then hydrolyze the acetal to obtain the diol intermediate.
- Convert the diol to an enose using iodine and triphenylphosphine, then couple with protected 5-fluorocytosine using NIS.
- Perform reductive dehalogenation with zinc and acetic acid, followed by base-catalyzed ester hydrolysis to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic sourcing perspective, this synthetic route offers compelling advantages that directly impact the bottom line and supply security. The transition from homogeneous liquid acids to heterogeneous resin catalysts simplifies the equipment requirements, as there is no need for specialized corrosion-resistant reactors or complex neutralization tanks. This reduction in capital expenditure (CAPEX) for plant modification makes the technology accessible for existing multipurpose facilities. Furthermore, the ability to telescope multiple steps without isolating intermediates significantly reduces the man-hours required for operation and lowers the consumption of utilities such as steam for distillation and electricity for vacuum pumps. These operational efficiencies translate into a more competitive pricing structure for the final intermediate, allowing pharmaceutical companies to allocate resources towards clinical development rather than excessive manufacturing costs.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the use of recyclable resin catalysts significantly lower raw material costs. By avoiding chromatographic purification in favor of crystallization and trituration, the process reduces the consumption of high-purity solvents and silica gel, which are major cost drivers in fine chemical production. The simplified workup procedures also decrease the volume of hazardous waste requiring disposal, leading to substantial savings in environmental compliance fees and waste management logistics.
- Enhanced Supply Chain Reliability: Starting from L-xylose, a commodity chemical derived from biomass, ensures a stable and sustainable raw material supply that is not subject to the geopolitical volatility often associated with petrochemical-derived starting materials. The robustness of the reaction conditions, which tolerate minor variations in temperature and stoichiometry without significant yield loss, ensures consistent batch-to-batch quality. This reliability minimizes the risk of production stoppages due to out-of-specification results, guaranteeing a continuous flow of material to downstream API manufacturers.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, with pilot plant data demonstrating successful execution in multi-kilogram batches. The reduction in solvent volume per kilogram of product and the avoidance of toxic reagents like heavy metals align with strict international environmental regulations such as REACH. This eco-friendly profile facilitates faster regulatory approval for the manufacturing site and enhances the corporate social responsibility (CSR) standing of the supply chain partners involved in the production of these life-saving antiviral agents.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its implementation. Common inquiries often revolve around the stability of the enose intermediate and the specific purity profiles achievable without column chromatography. The patent data provides clear guidance on storage conditions for sensitive intermediates, recommending low-temperature storage in specific solvents to prevent degradation. Additionally, the purification strategies employed, such as trituration with ethyl acetate or ethanol, are proven to remove key impurities like unreacted starting materials and side products effectively. The following answers address these technical considerations based on the experimental data provided in the patent documentation.
Q: What are the key advantages of the L-xylose route for Beta-L-FD4C synthesis?
A: The L-xylose route offers improved yields and purity compared to earlier methods. It utilizes cost-effective resin catalysts that are easily removed by filtration, reducing solid waste and simplifying downstream processing.
Q: How does this process address scalability concerns for antiviral intermediates?
A: The process is designed for mass production by avoiding evaporation to dryness between several steps. Telescoping reactions and solvent swaps allow for continuous processing, significantly enhancing throughput and operational efficiency.
Q: What purification methods are recommended for the final Beta-L-FD4C product?
A: The patent suggests trituration with solvents like ethyl acetate or ethanol, and crystallization. Avoiding silica gel chromatography is preferred for environmental reasons and cost reduction, relying instead on precipitation and washing steps.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Beta-L-FD4C Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-quality intermediates in the development of next-generation antiviral therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless. We adhere to stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify the identity and potency of every batch. Our commitment to excellence means that we do not just supply chemicals; we provide validated processes that meet the exacting standards of global regulatory bodies, ensuring your drug development pipeline remains uninterrupted.
We invite you to collaborate with us to leverage this advanced synthetic technology for your specific project needs. Our experts are ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements, demonstrating how our optimized processes can reduce your overall procurement spend. We encourage potential partners to contact our technical procurement team to request specific COA data and route feasibility assessments. By partnering with us, you gain access to a secure, scalable, and cost-effective supply chain solution that empowers you to bring vital medications to patients faster and more efficiently.