Advanced Tofacitinib Manufacturing: Overcoming Impurity Challenges with pH-Controlled Catalysis
Advanced Tofacitinib Manufacturing: Overcoming Impurity Challenges with pH-Controlled Catalysis
The pharmaceutical industry continuously seeks robust manufacturing processes that balance safety, cost-efficiency, and rigorous quality control, particularly for complex small-molecule inhibitors like Tofacitinib. A pivotal advancement in this domain is detailed in patent CN112094274A, which discloses an improved synthesis method specifically designed to mitigate the formation of difficult-to-remove impurities while eliminating the safety hazards associated with high-pressure hydrogenation. This technical breakthrough addresses a critical bottleneck in the production of JAK inhibitors, offering a pathway that enhances both the chemical purity of the active pharmaceutical ingredient (API) and the operational safety profile of the manufacturing facility. By integrating a precise pH-controlled hydrolysis step into the transfer hydrogenation workflow, this method effectively decomposes persistent formylated byproducts that have historically plagued scale-up efforts. For global procurement and R&D teams, understanding the nuances of this patented approach is essential for securing a reliable pharmaceutical intermediates supplier capable of delivering consistent, high-quality material without the baggage of legacy process limitations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of the key piperidine intermediate for Tofacitinib relied heavily on catalytic hydrogenation using molecular hydrogen gas under pressure. As illustrated in the traditional reaction pathways, this approach necessitates the use of specialized high-pressure reactors and stringent safety protocols to manage the explosive nature of hydrogen, creating significant capital expenditure barriers and operational risks for chemical manufacturers. Furthermore, alternative transfer hydrogenation methods utilizing formic acid or ammonium formate, while safer regarding pressure, introduced a different set of chemical challenges related to selectivity and impurity profiles. Specifically, the use of formic acid often leads to the formation of a stable N-formylated side product, referred to as Impurity A, which possesses physicochemical properties strikingly similar to the target molecule. This structural similarity makes the separation of Impurity A extremely difficult during purification, often requiring multiple recrystallizations or chromatographic steps that drastically reduce overall yield and increase production costs. Additionally, the presence of residual formic acid can complicate downstream processing, leading to potential degradation of the sensitive pyrrolo-pyrimidine core if the pH is not meticulously managed during workup.
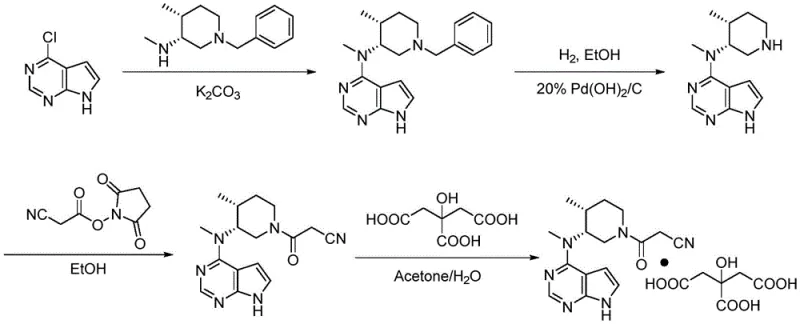
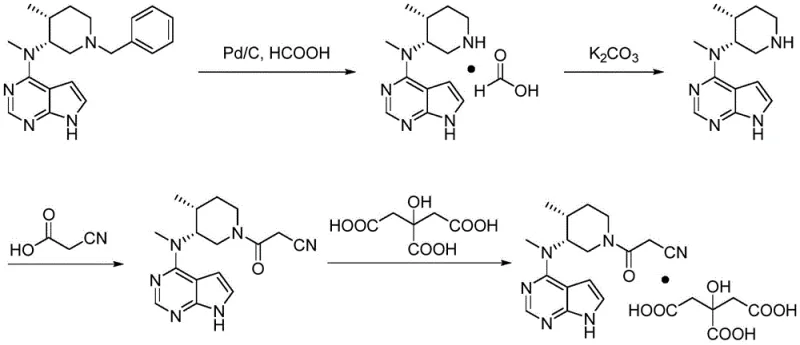
The Novel Approach
The innovative strategy presented in the patent data fundamentally alters the post-reaction workup to turn a chemical liability into a manageable variable. Instead of attempting to prevent the formation of Impurity A entirely during the reduction phase, the process accepts its transient formation and then actively destroys it through a controlled alkaline treatment. By adjusting the pH of the reaction mixture to a strongly alkaline level, specifically pH 13 or higher, immediately following the filtration of the palladium catalyst, the method triggers the rapid hydrolysis of the N-formyl bond in Impurity A. This clever manipulation converts the stubborn impurity back into the desired free amine (Compound II), effectively recycling the lost mass and boosting the overall yield of the intermediate. Simultaneously, this high pH environment converts any excess formic acid into its corresponding salt, such as sodium formate, which is highly water-soluble and easily removed during the subsequent aqueous extraction steps. This dual-action mechanism ensures that the isolated Compound II is of exceptional purity, setting a superior foundation for the final coupling reaction and significantly simplifying the purification burden for the final API.
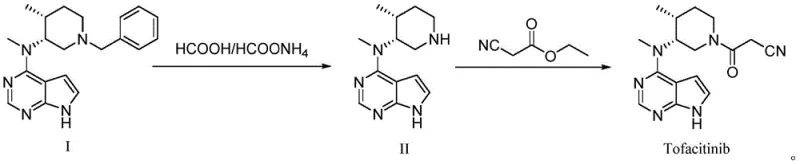
Mechanistic Insights into pH-Controlled Hydrolysis and Impurity Management
The core chemical innovation lies in the differential stability of the N-formyl group under varying pH conditions, a phenomenon that is exploited to purify the reaction stream in situ. Under neutral or acidic conditions, the N-formylated Impurity A is relatively stable, resisting hydrolysis and co-eluting with the product during standard workups. However, when the environment is shifted to a highly basic regime with a pH value greater than or equal to 13, the nucleophilic attack of hydroxide ions on the carbonyl carbon of the formyl group is dramatically accelerated. This base-catalyzed hydrolysis cleaves the amide bond, releasing the free secondary amine of the piperidine ring and generating formate ions. The patent data explicitly demonstrates that maintaining this alkaline condition is critical; experiments show that at pH 13, the content of Impurity A drops to undetectable levels, whereas at lower pH values like 12 or in acidic environments, the impurity persists or even remains intact. This mechanistic understanding allows process chemists to design a robust 'kill step' that guarantees the removal of this specific genotoxic or regulatory concern before the molecule proceeds to the next synthetic stage.
Beyond the primary hydrolysis mechanism, the process also optimizes the final isolation of Tofacitinib itself to prevent degradation. The final coupling reaction between Compound II and ethyl cyanoacetate is catalyzed by DBU, but the subsequent workup requires careful pH tuning to preserve product integrity. Data indicates that washing the organic phase with weakly alkaline water at a pH between 7 and 8.5 results in the lowest residual content of starting materials and the highest purity of the crude Tofacitinib. Deviating from this narrow window, either by making the solution too acidic or too alkaline, can lead to the hydrolysis of the nitrile group or other decomposition pathways that compromise the quality of the final drug substance. This precise control over the acid-base equilibrium throughout the synthesis underscores the sophistication of the method, ensuring that every unit operation contributes to the overall purity profile rather than introducing new contaminants.
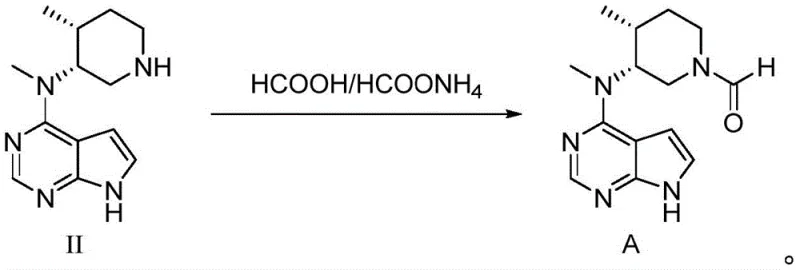
How to Synthesize Tofacitinib Intermediate Efficiently
The implementation of this improved synthesis route requires strict adherence to the specified reaction parameters to maximize the benefits of the pH-controlled purification strategy. The process begins with the reduction of the benzyl-protected precursor using ammonium formate or formic acid in a protic solvent like methanol, facilitated by a palladium catalyst at moderate temperatures ranging from 60°C to 70°C. Following the completion of the reduction, the critical intervention occurs: the reaction mixture is filtered to remove the catalyst, and the filtrate is immediately treated with a strong base, such as 30% sodium hydroxide solution, to raise the pH to 14. This step is not merely a neutralization but an active chemical transformation that decomposes impurities. After stirring under these alkaline conditions, the desired amine is extracted into an organic solvent, leaving the formate salts in the aqueous waste stream. The purified intermediate is then coupled with ethyl cyanoacetate in the presence of DBU to yield the final Tofacitinib structure, followed by a gentle wash with buffered water to ensure optimal crystal quality and purity.
- Perform debenzylation of Compound I using ammonium formate or formic acid with a Pd/C catalyst in methanol at 60-70°C.
- Filter the catalyst and adjust the filtrate pH to ≥13 using sodium hydroxide to hydrolyze formylated impurities back to the amine.
- Extract the purified Compound II and react with ethyl cyanoacetate using DBU catalyst to yield crude Tofacitinib.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this pH-controlled synthesis method offers substantial strategic advantages that extend beyond simple chemical yield improvements. The elimination of high-pressure hydrogen gas from the process removes the need for expensive, specialized autoclave infrastructure and reduces the regulatory burden associated with handling hazardous gases, thereby lowering the barrier to entry for contract manufacturing organizations (CMOs) and enhancing the resilience of the supply base. This shift allows for production in standard glass-lined or stainless steel reactors that are more readily available in the global chemical manufacturing network, reducing lead times for capacity booking and increasing the flexibility of production scheduling. Furthermore, the ability to convert impurities back into the desired product through the alkaline hydrolysis step represents a significant efficiency gain, as it minimizes the loss of valuable intermediates and reduces the volume of solvent and energy required for extensive purification procedures like column chromatography.
- Cost Reduction in Manufacturing: The process achieves cost optimization primarily through the simplification of the purification workflow and the improvement of overall mass balance. By chemically decomposing Impurity A back into the useful intermediate, the effective yield of the process is significantly increased without requiring additional raw material inputs, directly lowering the cost of goods sold (COGS) per kilogram of API. Additionally, the conversion of formic acid into water-soluble salts facilitates a cleaner extraction process, reducing the consumption of organic solvents and the associated costs of solvent recovery and waste disposal. The avoidance of high-pressure equipment also translates to lower capital depreciation costs and reduced maintenance expenses for the manufacturing facility, contributing to a more competitive pricing structure for the final pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: Utilizing liquid hydrogen donors like ammonium formate instead of compressed hydrogen gas mitigates the logistical risks associated with gas delivery and storage, which can be subject to transportation delays and regulatory restrictions. This change enhances the continuity of supply by relying on stable, solid, or liquid reagents that are easier to stockpile and handle in a standard warehouse environment. Moreover, the robustness of the pH-control step ensures consistent batch-to-batch quality, reducing the likelihood of failed batches due to impurity spikes that would otherwise disrupt delivery schedules. This reliability is crucial for pharmaceutical companies managing tight inventory levels and seeking to minimize the risk of drug shortages in the market.
- Scalability and Environmental Compliance: The method is inherently scalable because it avoids the mass transfer limitations often encountered in gas-liquid hydrogenation reactions at large scales, where mixing efficiency can impact reaction rates and safety. The liquid-phase transfer hydrogenation proceeds homogeneously or with suspended catalysts in a manner that is easily replicated from pilot plant to commercial tonnage production. From an environmental perspective, the process generates less hazardous waste by avoiding heavy metal scavengers often needed to remove leached catalysts from hydrogenation runs, and the aqueous waste stream containing formate salts is generally easier to treat biologically than complex organic solvent mixtures, aligning with modern green chemistry principles and regulatory expectations for sustainable manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this improved Tofacitinib synthesis method, drawing directly from the experimental data and process descriptions provided in the patent literature. Understanding these details is vital for technical teams evaluating the feasibility of technology transfer or assessing the quality standards of potential suppliers. The answers reflect the specific operational parameters, such as pH thresholds and temperature ranges, that define the success of this manufacturing route.
Q: How does the pH adjustment step eliminate Impurity A in Tofacitinib synthesis?
A: By adjusting the reaction mixture to a highly alkaline environment (pH ≥ 13) after the reduction step, the stable formylated impurity (Impurity A) undergoes rapid hydrolysis, reverting back to the desired amine (Compound II) while converting residual formic acid into water-soluble salts that are easily removed during extraction.
Q: Why is this method safer than traditional hydrogenation routes?
A: Traditional methods require high-pressure hydrogen gas (H2) which poses significant explosion risks and requires specialized autoclave equipment. This improved method utilizes ammonium formate or formic acid as a liquid hydrogen source under atmospheric pressure, drastically reducing operational hazards and equipment costs.
Q: What is the optimal pH range for the final extraction of Tofacitinib?
A: During the final workup after coupling with ethyl cyanoacetate, washing with weakly alkaline water at pH 7-8.5 is critical. This specific range minimizes the degradation of Tofacitinib while effectively removing unreacted starting materials and acidic byproducts.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tofacitinib Supplier
As the demand for JAK inhibitors continues to grow globally, partnering with a manufacturer that possesses deep expertise in advanced intermediate synthesis is critical for maintaining a competitive edge in the pharmaceutical market. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to deliver high-value intermediates with unmatched consistency. Our technical team is fully versed in the nuances of pH-controlled catalytic processes and impurity management strategies, ensuring that every batch of Tofacitinib intermediate meets stringent purity specifications and rigorous QC labs standards. We understand that the transition from laboratory scale to industrial manufacturing requires not just chemical knowledge but also engineering excellence, which is why our facilities are equipped to handle complex multi-step syntheses with the precision required for regulatory compliance.
We invite procurement leaders and R&D directors to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By collaborating with our technical procurement team, you can access specific COA data and route feasibility assessments that demonstrate how our optimized manufacturing processes can enhance your supply chain efficiency. Whether you require clinical trial materials or commercial-scale API intermediates, NINGBO INNO PHARMCHEM is committed to providing the reliability, transparency, and technical support necessary to accelerate your drug development timelines and secure your market position.