Revolutionizing Canertinib Manufacturing: A Scalable and Economic Synthetic Route
The pharmaceutical landscape for oncology treatments continues to evolve, driven by the demand for more efficient manufacturing processes for potent kinase inhibitors like Canertinib. Patent CN103242244A introduces a transformative preparation method that addresses critical bottlenecks in the existing supply chain for this irreversible ErbB family inhibitor. Unlike traditional pathways that rely on complex nitro-intermediates and harsh halogenation steps, this novel approach leverages a strategically selected amino-hydroxy precursor to streamline the synthetic sequence. By fundamentally rethinking the order of functional group installation, the process achieves a remarkable balance between atom economy and operational simplicity. This technical breakthrough is particularly significant for global supply chains seeking reliable pharmaceutical intermediates supplier partnerships that can guarantee continuity without compromising on quality or environmental standards. The shift away from difficult-to-source fluorochemicals towards commodity-grade starting materials represents a paradigm shift in how we approach the commercial scale-up of complex kinase inhibitors.
The Limitations of Conventional Methods vs. The Novel Approach
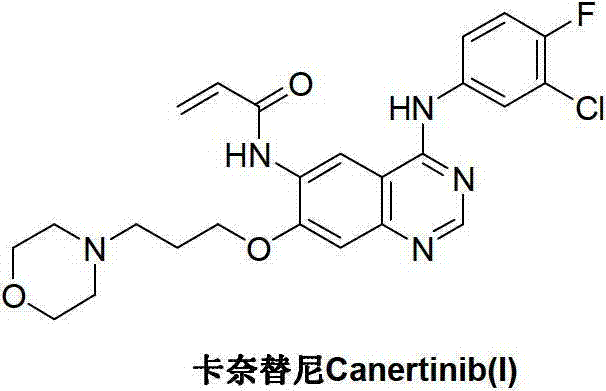
The Limitations of Conventional Methods
Historically, the industrial synthesis of Canertinib has been plagued by reliance on routes originating from 4-[(3-chloro-4-fluorophenyl)amino]-6-nitro-7-fluoroquinazoline derivatives. As illustrated in the prior art reaction scheme, these conventional methods necessitate a multi-step sequence involving nitration, chlorination, and subsequent nucleophilic substitution under alkaline conditions. 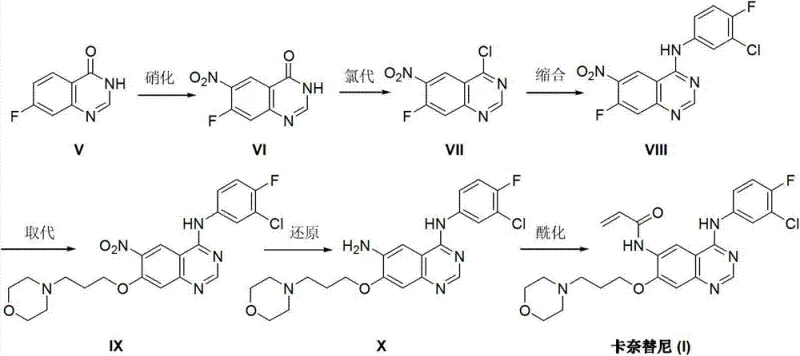 The inherent drawbacks of this legacy chemistry are manifold; firstly, the starting fluorochemicals are often expensive and subject to volatile market availability, creating supply chain vulnerabilities. Secondly, the presence of the nitro group necessitates a subsequent reduction step, which frequently generates difficult-to-remove impurities and requires rigorous purification protocols, often involving column chromatography. Such purification techniques are notoriously difficult to translate from the laboratory to multi-ton manufacturing scales, leading to significant yield losses and increased waste generation. Furthermore, the handling of nitro intermediates poses safety risks related to thermal stability, adding another layer of complexity to process safety management in large-scale reactors.
The inherent drawbacks of this legacy chemistry are manifold; firstly, the starting fluorochemicals are often expensive and subject to volatile market availability, creating supply chain vulnerabilities. Secondly, the presence of the nitro group necessitates a subsequent reduction step, which frequently generates difficult-to-remove impurities and requires rigorous purification protocols, often involving column chromatography. Such purification techniques are notoriously difficult to translate from the laboratory to multi-ton manufacturing scales, leading to significant yield losses and increased waste generation. Furthermore, the handling of nitro intermediates poses safety risks related to thermal stability, adding another layer of complexity to process safety management in large-scale reactors.
The Novel Approach
In stark contrast, the methodology disclosed in CN103242244A proposes a streamlined three-step sequence that bypasses these historical hurdles entirely. The new strategy initiates with 6-amino-7-hydroxy-3,4-dihydroquinazoline-4-one, a scaffold that allows for the direct installation of the morpholine-propoxy side chain via etherification. 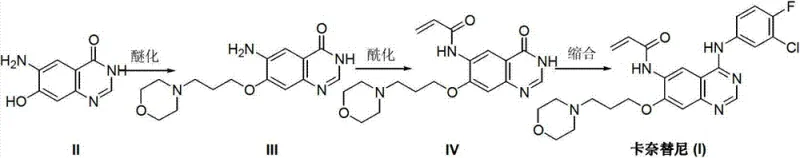 This inversion of the synthetic logic eliminates the need for the problematic nitro-reduction step, thereby inherently improving the impurity profile of the intermediate stream. The subsequent acylation and condensation steps are designed to be robust and scalable, utilizing standard peptide coupling chemistry that is well-understood in process development. By removing the dependency on scarce fluorinated building blocks at the early stages and avoiding chromatographic purification, this route offers a clear pathway for cost reduction in API manufacturing. The simplicity of the work-up procedures, which rely on pH adjustments and crystallization rather than complex separations, underscores the process's suitability for industrialization and its alignment with green chemistry principles.
This inversion of the synthetic logic eliminates the need for the problematic nitro-reduction step, thereby inherently improving the impurity profile of the intermediate stream. The subsequent acylation and condensation steps are designed to be robust and scalable, utilizing standard peptide coupling chemistry that is well-understood in process development. By removing the dependency on scarce fluorinated building blocks at the early stages and avoiding chromatographic purification, this route offers a clear pathway for cost reduction in API manufacturing. The simplicity of the work-up procedures, which rely on pH adjustments and crystallization rather than complex separations, underscores the process's suitability for industrialization and its alignment with green chemistry principles.
Mechanistic Insights into Mitsunobu Etherification and Amide Coupling
The cornerstone of this improved synthesis lies in the initial etherification step, which employs Mitsunobu reaction conditions to couple the phenolic hydroxyl group of the quinazoline core with 3-(4-morpholinyl)-1-propanol. Mechanistically, this involves the activation of the alcohol by triphenylphosphine and a diazodicarboxylate (such as DIAD), creating a highly reactive alkoxyphosphonium intermediate that is subsequently displaced by the phenoxide nucleophile. This specific transformation is highly regioselective for the 7-position hydroxyl group over the 6-position amino group, ensuring the correct structural orientation without the need for extensive protecting group strategies. The choice of tetrahydrofuran (THF) as the solvent facilitates the solubility of both the heterocyclic substrate and the phosphine reagents, while maintaining a reaction temperature at room temperature minimizes thermal degradation of sensitive functional groups. This mechanistic precision is critical for R&D directors focused on purity, as it prevents the formation of regio-isomers that could complicate downstream processing.
Following the etherification, the process moves to the installation of the acrylamide warhead and the final aniline condensation. The acylation of the 6-amino group with acryloyl chloride is conducted under mild basic conditions to scavenge the generated HCl, preventing acid-catalyzed polymerization of the acrylic double bond. The final condensation step, linking the quinazoline ketone intermediate with 4-fluoro-3-chloroaniline, utilizes advanced coupling agents like BOP or HBTU. These reagents activate the carbonyl species effectively, allowing the nucleophilic attack by the aniline nitrogen to proceed efficiently even at moderate temperatures of 50-60°C. The use of non-nucleophilic bases such as DBU or DBN further drives the equilibrium towards product formation while minimizing side reactions. This careful selection of reagents ensures that the final cyclization and bond formation occur with high fidelity, resulting in a product that meets stringent purity specifications without the need for exhaustive purification.
How to Synthesize Canertinib Efficiently
The practical execution of this synthesis involves a logical progression of unit operations that are easily adaptable to standard stainless steel reactor trains used in fine chemical manufacturing. The process begins with the preparation of the ether-linked intermediate, followed by the introduction of the acrylamide functionality, and concludes with the final coupling of the aniline moiety. Each step has been optimized to maximize yield while minimizing solvent usage and waste generation, reflecting a deep understanding of process chemistry. For technical teams looking to implement this route, the detailed standardized synthetic steps provided below offer a robust framework for technology transfer and pilot plant trials. The clarity of the reaction conditions, including specific molar ratios and temperature ranges, reduces the risk of batch failure and accelerates the timeline from lab bench to commercial production.
- Perform etherification of 6-amino-7-hydroxy-3,4-dihydroquinazoline-4-one with 3-(4-morpholinyl)-1-propanol using Mitsunobu conditions.
- Conduct acylation of the resulting amino-intermediate with acryloyl chloride to form the acrylamido derivative.
- Execute condensation with 4-fluoro-3-chloroaniline using peptide coupling agents like BOP or HBTU to finalize the Canertinib structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the shift to this new synthetic route offers substantial strategic benefits that extend beyond simple unit cost calculations. The primary advantage lies in the sourcing of raw materials; by utilizing 6-amino-7-hydroxy-3,4-dihydroquinazoline-4-one, manufacturers can access commodity chemicals that are widely available from multiple global suppliers, thereby reducing single-source dependency risks. This diversification of the supply base enhances supply chain reliability and provides greater leverage in price negotiations, ensuring long-term stability for production planning. Furthermore, the elimination of column chromatography—a resource-intensive and low-throughput purification method—drastically reduces the consumption of silica gel and organic solvents, leading to significant operational expenditure savings. These efficiencies translate directly into a more competitive cost structure, allowing for better margin management in a pricing-sensitive generic and biosimilar market environment.
- Cost Reduction in Manufacturing: The streamlined nature of this three-step process inherently lowers manufacturing costs by reducing the total number of unit operations and the associated labor and utility requirements. By avoiding the use of expensive and hazardous fluorochemicals in the early stages, the raw material bill of materials is significantly optimized. Additionally, the ability to purify intermediates through crystallization and extraction rather than chromatography reduces solvent recovery costs and waste disposal fees. These cumulative effects result in a leaner manufacturing process that delivers substantial cost savings without compromising the quality of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The reliance on readily available starting materials mitigates the risk of supply disruptions that often plague specialized fluorochemical supply chains. Since the key precursors are simpler heterocycles and common alcohols, lead times for raw material procurement can be significantly shortened, enabling more responsive production scheduling. This agility is crucial for meeting the fluctuating demands of the pharmaceutical market, ensuring that inventory levels can be maintained consistently to support continuous drug product manufacturing. The robustness of the chemistry also means that batch-to-batch variability is minimized, further stabilizing the supply of high-quality intermediates.
- Scalability and Environmental Compliance: The process is explicitly designed with industrial scalability in mind, avoiding reactions that are difficult to control exothermically or those that generate excessive toxic waste. The absence of nitration steps removes the generation of nitro-containing wastewater, simplifying effluent treatment and ensuring compliance with increasingly strict environmental regulations. The use of standard solvents like THF, acetonitrile, and ethyl acetate facilitates efficient solvent recovery and recycling, aligning the process with green chemistry initiatives. This environmental compatibility not only reduces regulatory burden but also enhances the corporate sustainability profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Canertinib synthesis route. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a factual basis for decision-making. Understanding these nuances is essential for stakeholders evaluating the feasibility of adopting this technology for their own production portfolios. The answers reflect a commitment to transparency and technical accuracy, ensuring that all parties have a clear understanding of the process capabilities and limitations.
Q: What are the primary advantages of the new Canertinib synthesis route over conventional methods?
A: The new route utilizes readily available 6-amino-7-hydroxy-3,4-dihydroquinazoline-4-one as a starting material, avoiding the use of scarce fluorochemicals and hazardous nitration steps required in traditional pathways. This significantly simplifies purification by eliminating the need for column chromatography, making it highly suitable for industrial scale-up.
Q: Which coupling agents are recommended for the final condensation step?
A: The patent specifies the use of efficient peptide coupling agents such as benzotriazole-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) or HBTU. These reagents, combined with bases like DBU or DBN in solvents like acetonitrile, ensure high conversion rates and manageable reaction temperatures between 50-60°C.
Q: How does this process impact the purity profile of the final API intermediate?
A: By avoiding the reduction of nitro groups found in older methods, this route minimizes the formation of associated reduction byproducts and azo-impurities. The use of crystallization for purification in the final steps, rather than chromatography, supports the production of high-purity intermediates consistent with stringent pharmaceutical standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Canertinib Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic routes to maintain competitiveness in the global pharmaceutical market. Our team of expert process chemists has extensively evaluated the methodology described in CN103242244A and possesses the technical capability to execute this route with precision and efficiency. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from pilot scale to full manufacturing is seamless and risk-mitigated. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Canertinib intermediate meets the highest international standards for safety and efficacy.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis can benefit your specific supply chain needs. By collaborating with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic advantages of switching to this greener, more efficient process. We encourage you to request specific COA data and route feasibility assessments to validate the performance of our materials in your downstream applications. Together, we can drive innovation and efficiency in the production of life-saving oncology therapies.