Advanced Acid-Catalyzed Cyclization for High-Purity Fluorene Pharmaceutical Intermediates Manufacturing
Advanced Acid-Catalyzed Cyclization for High-Purity Fluorene Pharmaceutical Intermediates Manufacturing
The landscape of medicinal chemistry continuously demands more efficient routes to complex scaffolds, particularly those containing the 3-(9-hydrofluoren-9-yl)propionyl motif which has demonstrated significant biological activity against mammalian ribonucleotide reductase and bacterial targets. Patent CN110256247B, published in September 2021, introduces a transformative synthetic methodology for producing 2-((9-hydrofluoren-9-yl)methyl)malonate, a versatile precursor that can be readily hydrolyzed and decarboxylated to access these valuable propionic acid derivatives. This innovation shifts the paradigm from traditional, low-yielding alkylation strategies to a highly atom-economical intramolecular ring-opening arylation of 2-biarylcyclopropyl-1,1-dicarboxylates. By leveraging strong acid catalysis under mild conditions, this process addresses critical pain points in impurity control and operational complexity, positioning it as a superior choice for reliable pharmaceutical intermediate supplier networks seeking to optimize their API manufacturing pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historical approaches to constructing the fluorene-propionate skeleton have been plagued by inherent inefficiencies and poor selectivity. As illustrated in the prior art, early attempts by Smith et al. utilized 9-hydrofluorene and ethyl acrylate in the presence of tetrabutylammonium fluoride, resulting in a dismal 4% yield accompanied by significant formation of bis-addition byproducts.
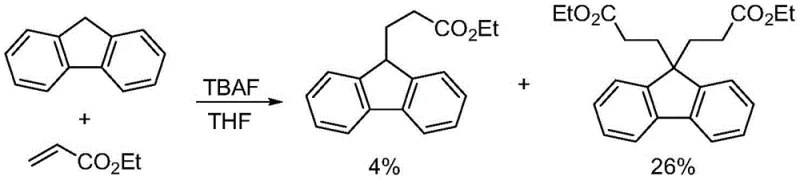
Similarly, Ulijn et al. employed sodium hydride to react 9-hydrofluorene with methyl 3-bromopropionate, yet this method also suffered from suboptimal efficiency, delivering only a 20% yield. Alternative strategies involving Wittig olefination followed by palladium-catalyzed hydrogenation, while avoiding multiple additions, introduce substantial economic and environmental burdens due to the generation of large amounts of phosphine oxide waste and the requirement for expensive transition metal removal steps. These conventional pathways often necessitate harsh conditions, extensive purification protocols, and result in unacceptable material loss, making them ill-suited for cost reduction in pharmaceutical intermediate manufacturing.
The Novel Approach
In stark contrast, the methodology disclosed in CN110256247B utilizes a strategic intramolecular cyclization of a pre-functionalized cyclopropane precursor. The core transformation involves treating a 2-biarylcyclopropyl-1,1-dicarboxylate (Formula I) with an excess of trifluoromethanesulfonic acid in dichloromethane at room temperature. This elegant cascade reaction simultaneously opens the strained cyclopropane ring and closes the five-membered fluorene ring in a single operational step.
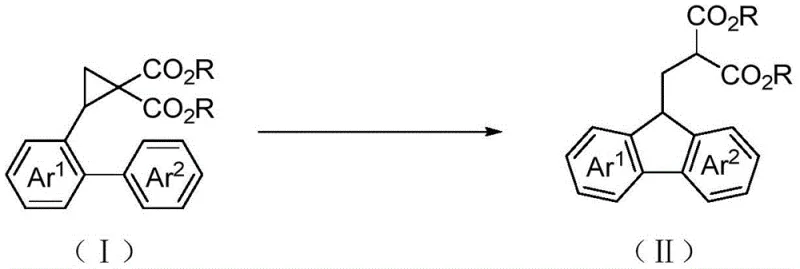
This novel approach eliminates the need for transition metals entirely and operates under exceptionally mild conditions, typically completing within 1 hour. The process boasts remarkable versatility, accommodating a wide array of substituents on the biaryl system, including methyl, methoxy, hydroxyl, halogens, and even heteroaryl rings like thiophene and indole. By shifting the bond-forming strategy from intermolecular alkylation to intramolecular electrophilic aromatic substitution facilitated by ring strain release, the invention achieves isolated yields ranging from 71% to 98%, representing a quantum leap in synthetic efficiency and process reliability.
Mechanistic Insights into Trifluoromethanesulfonic Acid-Catalyzed Cyclization
The success of this transformation hinges on the unique reactivity of the donor-acceptor cyclopropane moiety under superacidic conditions. Upon exposure to trifluoromethanesulfonic acid (TfOH), the electron-rich cyclopropane ring undergoes protonation or Lewis acid coordination, significantly enhancing the electrophilicity of the ring carbons. This activation triggers a regioselective ring-opening event, generating a stabilized benzylic carbocation intermediate. The pendant aryl ring, positioned ideally for intramolecular attack, then acts as a nucleophile, engaging in an electrophilic aromatic substitution (Friedel-Crafts type) to close the central five-membered ring of the fluorene system. The presence of two ester groups at the 1-position of the cyclopropane serves a dual purpose: it stabilizes the developing negative charge during the initial enolate-like character or stabilizes the cationic intermediate through resonance, and it provides the necessary malonate functionality for downstream derivatization.
From an impurity control perspective, this mechanism offers distinct advantages over radical or metal-catalyzed pathways. The intramolecular nature of the cyclization inherently suppresses intermolecular oligomerization or polymerization side reactions that often plague intermolecular alkylations. Furthermore, the use of a homogeneous strong acid catalyst ensures uniform reaction kinetics, minimizing the formation of regioisomers. The patent data confirms that electron-donating groups on the attacking aryl ring (such as methoxy or methyl) facilitate the cyclization through increased nucleophilicity, while electron-withdrawing groups (like nitro or trifluoromethyl) are still well-tolerated, albeit sometimes requiring slightly higher acid equivalents (up to 10 equivalents) to drive the reaction to completion. This robustness ensures a clean impurity profile, simplifying the subsequent isolation and purification steps required for high-purity pharmaceutical intermediate production.
How to Synthesize 2-((9-Hydrofluoren-9-yl)methyl)malonate Efficiently
The synthesis protocol outlined in the patent is designed for operational simplicity and scalability, beginning with the preparation of the cyclopropane precursor via a Knoevenagel condensation followed by a Corey-Chaykovsky reaction. Once the precursor (Formula I) is secured, the critical cyclization step is performed under strictly anhydrous conditions to prevent hydrolysis of the sensitive intermediates. The reaction mixture is quenched carefully to manage the exotherm associated with neutralizing the strong acid, followed by a standard aqueous workup that effectively removes acidic residues and polar byproducts. For detailed standardized operating procedures regarding stoichiometry, addition rates, and specific workup parameters, please refer to the guide below.
- Preparation of the cyclopropane precursor (Formula I) via Knoevenagel condensation of 2-biphenylcarboxaldehyde with diethyl malonate, followed by Corey-Chaykovsky cyclopropanation.
- Dissolution of the precursor in dichloromethane under nitrogen atmosphere at room temperature.
- Addition of 1 to 10 molar equivalents of trifluoromethanesulfonic acid, stirring for 1 hour, followed by neutralization and extraction to isolate the target fluorene derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this acid-catalyzed cyclization route offers compelling strategic benefits that extend beyond mere chemical yield. The elimination of precious metal catalysts such as palladium, which are required in the hydrogenation steps of older Wittig-based routes, directly translates to significant cost reduction in API manufacturing. Removing the need for expensive metal scavengers and the associated validation testing for residual heavy metals streamlines the quality control workflow and reduces the overall cost of goods sold. Additionally, the reagents employed—dichloromethane and trifluoromethanesulfonic acid—are commodity chemicals with stable global supply chains, mitigating the risk of raw material shortages that often affect specialized organometallic reagents.
- Cost Reduction in Manufacturing: The process achieves substantial cost savings by utilizing cheap, commercially available starting materials and avoiding the high capital expenditure associated with high-pressure hydrogenation equipment. The reaction proceeds at room temperature, eliminating the energy costs linked to heating or cryogenic cooling, and the simple extractive workup reduces solvent consumption and waste disposal fees compared to chromatography-heavy purifications required for lower-selectivity methods.
- Enhanced Supply Chain Reliability: By relying on robust organic transformations rather than sensitive catalytic cycles, the manufacturing process becomes less susceptible to batch-to-batch variability caused by catalyst deactivation or ligand degradation. The broad substrate scope means that a single platform technology can be used to produce a diverse library of substituted fluorene derivatives, allowing for flexible inventory management and faster response times to changing market demands for specific analogues.
- Scalability and Environmental Compliance: The patent explicitly validates the method on gram scales with near-quantitative yields, demonstrating its readiness for commercial scale-up of complex pharmaceutical intermediates. The absence of toxic heavy metals and the use of standard organic solvents simplify regulatory compliance and environmental, health, and safety (EHS) assessments, facilitating smoother technology transfer from R&D to pilot and production plants.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis technology. These answers are derived directly from the experimental data and embodiments provided in the patent documentation, ensuring accuracy for process development teams evaluating this route for potential licensing or contract manufacturing opportunities.
Q: What are the primary advantages of this acid-catalyzed method over traditional alkylation?
A: Unlike traditional methods using 9-hydrofluorene which suffer from low yields (4-20%) and poly-alkylation issues, this novel cyclization approach achieves yields up to 98% with excellent regioselectivity and avoids expensive transition metal catalysts.
Q: Is this process scalable for industrial production?
A: Yes, the patent explicitly demonstrates reliability on gram-scale preparations (e.g., 10 mmol scale in Example 2) with simple workup procedures involving neutralization and extraction, indicating strong potential for commercial scale-up.
Q: What is the functional group tolerance of this cyclization reaction?
A: The method exhibits broad substrate scope, successfully tolerating electron-donating groups (methyl, methoxy), electron-withdrawing groups (nitro, halogen, trifluoromethyl), and heteroaryl systems like thiophene and indole.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-((9-Hydrofluoren-9-yl)methyl)malonate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in accelerating drug discovery and development timelines. Our technical team has thoroughly analyzed the pathway described in CN110256247B and confirmed its viability for large-scale production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of fluorene derivative delivered meets the exacting standards required for clinical and commercial applications.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next project. By partnering with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in acid-catalyzed cyclizations can drive efficiency and reliability in your supply chain.