Advanced Two-Step Synthesis of Belinostat Intermediates for Commercial Scale-Up
Advanced Two-Step Synthesis of Belinostat Intermediates for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust synthetic pathways for oncology drug candidates, particularly for histone deacetylase inhibitors like Belinostat which are currently in advanced clinical stages. Patent CN103724239A introduces a groundbreaking preparation method for the critical intermediate 3-(3-phenylamidosulfonyl-phenyl)-acrylate, addressing long-standing inefficiencies in existing manufacturing protocols. This technical disclosure outlines a concise two-step synthesis that significantly minimizes side reactions while achieving superior yields that meet rigorous industrial production standards. By shifting away from convoluted multi-step sequences, this innovation provides a reliable Belinostat intermediate supplier pathway that ensures consistent quality and availability for downstream API synthesis. The strategic implementation of this technology allows pharmaceutical manufacturers to secure their supply chains against the volatility associated with complex chemical manufacturing processes.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this key pharmaceutical intermediate has relied on arduous routes such as the one described in WO230879, which involves a lengthy sequence starting from benzaldehyde and proceeding through sulfonation and Knoevenagel condensation steps. These traditional methodologies are plagued by excessive reaction steps that accumulate impurities at each stage, resulting in a cumbersome aftertreatment process that drastically reduces the ultimate yield of the target molecule. The reliance on harsh reagents and multiple isolation procedures not only increases the operational complexity but also elevates the cost burden due to significant material loss and extended processing times. Furthermore, the generation of substantial waste streams from these inefficient routes poses environmental compliance challenges that modern green chemistry initiatives strive to eliminate from the supply chain. Consequently, manufacturers face difficulties in scaling these processes without compromising on purity or economic viability.
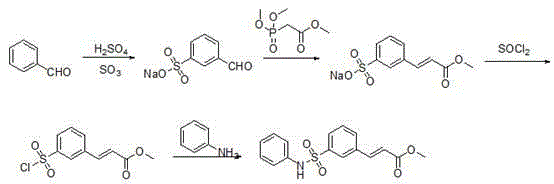
The Novel Approach
In stark contrast to the legacy methods, the novel approach disclosed in the patent utilizes a streamlined strategy that constructs the core sulfonamide structure early in the synthesis before introducing the acrylate moiety via a efficient coupling reaction. This methodology leverages the reactivity of bromobenzene sulfonyl chloride and aniline to form a stable intermediate, which then undergoes a palladium-catalyzed transformation to install the necessary unsaturated ester functionality with high precision. The reduction in step count directly correlates to a simplification of the workup procedure, allowing for easier purification through standard crystallization techniques rather than complex chromatographic separations. By optimizing the reaction conditions to favor the desired product formation, this route achieves yields that are substantially higher than previous iterations, thereby enhancing the overall material throughput for cost reduction in pharmaceutical intermediates manufacturing. This shift represents a paradigm change in how high-value oncology intermediates are produced commercially.
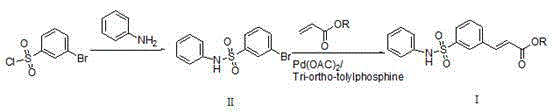
Mechanistic Insights into Palladium-Catalyzed Heck Coupling
The core of this technological advancement lies in the sophisticated application of palladium-catalyzed cross-coupling chemistry, specifically a Heck-type reaction that joins the sulfonamide intermediate with an acrylate ester under controlled thermal conditions. The catalytic cycle initiates with the oxidative addition of the palladium species into the carbon-bromine bond of the sulfonamide, a critical step that is facilitated by the electron-withdrawing nature of the sulfonyl group which activates the aromatic ring. Subsequent coordination and insertion of the acrylate double bond into the palladium-carbon bond proceed with high regioselectivity, ensuring that the trans-isomer is predominantly formed as required for the biological activity of the final drug substance. The presence of tri-o-tolylphosphine as a ligand stabilizes the active catalytic species and prevents the aggregation of palladium black, which is a common deactivation pathway in such transformations. This mechanistic precision ensures that the reaction proceeds to completion with minimal formation of homocoupling byproducts or debrominated species that would otherwise complicate the purification landscape.
Impurity control is meticulously managed through the selection of specific acid binding agents and solvents that suppress side reactions such as hydrolysis or polymerization of the acrylate component during the heating phase. The patent specifies the use of diisopropylethylamine or triethylamine in toluene at approximately 80°C, conditions that are optimized to balance reaction kinetics with thermal stability of the sensitive functional groups present in the molecule. Post-reaction processing involves a strategic hot filtration step to remove insoluble palladium residues and amine salts, followed by acid washing to eliminate any unreacted basic components that could affect the stability of the final solid. The final recrystallization from ethanol serves as a powerful purification tool, leveraging the solubility differences between the target acrylate and any remaining organic impurities to deliver a product with exceptional chemical purity. This rigorous control over the chemical environment ensures that the intermediate meets the stringent specifications required for subsequent API manufacturing steps.
How to Synthesize 3-(3-phenylamidosulfonyl-phenyl)-acrylate Efficiently
Implementing this synthesis requires careful attention to the stoichiometry and addition rates of reagents to maintain the exothermic profile within safe operating limits while maximizing conversion efficiency. The process begins with the formation of the sulfonamide intermediate in a non-protonic solvent like tetrahydrofuran, where the temperature is carefully maintained around 40°C to prevent over-reaction or degradation of the starting materials. Once the intermediate is isolated and characterized, it is subjected to the coupling conditions using a precise loading of palladium catalyst and phosphine ligand to ensure consistent batch-to-batch performance. Detailed standardized synthetic steps see the guide below for specific parameters regarding mixing speeds, addition times, and crystallization cooling rates that are critical for reproducibility. Adhering to these optimized protocols allows production teams to achieve the high yields reported in the patent data while maintaining a safe and controlled manufacturing environment.
- Perform nucleophilic substitution between bromobenzene sulfonyl chloride and aniline in a non-protonic solvent like THF with a catalyst such as DMAP or PPY at approximately 40°C to form the sulfonamide intermediate.
- Execute a palladium-catalyzed coupling reaction between the sulfonamide intermediate and an acrylate ester using tri-o-tolylphosphine ligand and an acid binding agent in toluene at 80°C.
- Purify the crude product through hot filtration, acid washing, and recrystallization from ethanol to achieve high purity specifications suitable for pharmaceutical applications.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this streamlined synthetic route offers profound advantages in terms of cost structure and logistical reliability compared to traditional sourcing models. By eliminating the need for multiple intermediate isolations and complex purification trains, the manufacturing overhead is significantly reduced, translating into more competitive pricing structures for long-term supply agreements without compromising on quality standards. The simplified process flow also reduces the dependency on specialized equipment for hazardous transformations, thereby lowering the barrier for qualified suppliers to enter the market and increasing the resilience of the supply base against disruptions. Furthermore, the high yield and purity profile minimize the risk of batch failures, ensuring that delivery schedules are met consistently and reducing the need for safety stock inventory that ties up working capital. This operational efficiency supports a more agile supply chain capable of responding rapidly to the fluctuating demands of clinical and commercial drug production cycles.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts in the initial step and the reduction of overall unit operations lead to substantial cost savings by decreasing solvent consumption and waste disposal fees associated with complex chemical processing. The high atom economy of the coupling reaction ensures that raw material costs are optimized, as a greater proportion of the input mass is converted into valuable product rather than lost as byproduct waste. Additionally, the ability to use commodity chemicals like aniline and acrylates in a direct fashion avoids the premium pricing often associated with highly functionalized building blocks required by older synthetic routes. These cumulative efficiencies create a robust economic model that supports sustainable pricing strategies for high-purity pharmaceutical intermediates in a competitive global market.
- Enhanced Supply Chain Reliability: The use of readily available starting materials and standard reaction conditions mitigates the risk of raw material shortages that often plague specialty chemical supply chains dependent on obscure reagents. The robustness of the two-step process allows for flexible manufacturing scheduling, enabling suppliers to scale production up or down based on real-time demand signals without extensive requalification efforts. This flexibility ensures reducing lead time for high-purity pharmaceutical intermediates, allowing drug developers to accelerate their timelines from clinical trials to commercial launch with confidence in their material supply. The consistency of the process also simplifies quality auditing, fostering stronger partnerships between chemical suppliers and pharmaceutical innovators.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up of complex pharmaceutical intermediates in mind, utilizing solvents and conditions that are easily managed in large-scale reactor systems without requiring exotic high-pressure or cryogenic equipment. The reduction in waste generation aligns with increasingly strict environmental regulations, reducing the regulatory burden and potential liability associated with hazardous waste management in chemical manufacturing facilities. The simplified workup procedure minimizes the volume of aqueous waste streams, lowering the cost and complexity of wastewater treatment operations. This environmentally conscious approach not only meets compliance requirements but also enhances the corporate sustainability profile of the manufacturing organization.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and specification of this critical oncology intermediate based on the patented technology. Understanding these details helps stakeholders evaluate the feasibility of integrating this material into their development pipelines and ensures alignment on quality expectations. The answers are derived directly from the experimental data and process descriptions provided in the intellectual property documentation to ensure accuracy and relevance. Stakeholders are encouraged to review these insights to better understand the value proposition of this advanced manufacturing approach.
Q: What are the primary impurities in conventional Belinostat intermediate synthesis?
A: Conventional routes often suffer from complex byproduct profiles due to multi-step sequences involving harsh sulfonation and condensation conditions, leading to difficult purification and lower overall yields compared to the direct coupling method.
Q: How does the new palladium-catalyzed route improve scalability?
A: By reducing the synthesis to two robust steps with high conversion rates and simplified workup procedures involving standard crystallization, the process eliminates bottleneck operations and enhances suitability for large-scale industrial production.
Q: Is the palladium catalyst recoverable in this process?
A: While the patent focuses on high conversion efficiency, the use of heterogeneous filtration steps during workup allows for the potential removal of metal residues, ensuring the final product meets stringent heavy metal specifications required for API manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-(3-phenylamidosulfonyl-phenyl)-acrylate Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production for global pharmaceutical clients. Our technical team is adept at adapting patented routes like CN103724239A to fit specific GMP requirements, ensuring that every batch meets stringent purity specifications and rigorous QC labs standards. We understand the critical nature of oncology supply chains and are committed to providing uninterrupted supply through our robust quality management systems and redundant manufacturing capabilities. Partnering with us means gaining access to deep chemical expertise that can troubleshoot process deviations and optimize yields further to drive down your overall cost of goods sold.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements and project timelines. Our experts are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your drug development goals. By collaborating early in the development phase, we can ensure a seamless transition from clinical supply to commercial manufacturing, securing your position in the competitive oncology market. Reach out today to discuss how we can become your strategic partner for high-quality chemical intermediates.