Advanced Acid-Catalyzed Synthesis of 3,7-Dimethyl-6-Carbonyl-2-Octenal for Commercial Scale-Up
The global demand for high-purity norisoprenoid derivatives, particularly those serving as critical intermediates in the flavor, fragrance, and pharmaceutical sectors, continues to drive innovation in synthetic methodology. A pivotal advancement in this domain is detailed in patent CN107032971B, which discloses a highly efficient preparation method for 3,7-dimethyl-6-carbonyl-2-octenal. This specific compound, a C13-norisoprenoid and citral derivative, holds immense value due to its unique olfactory profile and utility as a versatile building block for complex organic synthesis. The patented technology represents a significant departure from legacy synthetic routes, shifting away from hazardous and energy-intensive processes toward a streamlined, acid-catalyzed hydrolysis performed under ambient conditions. By utilizing a medium-concentration sulfuric acid system, the process achieves remarkable selectivity and yield without the need for cryogenic cooling or exotic catalysts. For R&D directors and procurement strategists alike, this development signals a potential paradigm shift in how high-value aroma chemicals and pharmaceutical intermediates are sourced, offering a pathway to reduce both operational expenditure and supply chain volatility.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3,7-dimethyl-6-carbonyl-2-octenal has been plagued by significant technical and economic hurdles that hindered its widespread commercial adoption. Early literature, such as the work by Carl G et al. in 1989, relied heavily on the ozonolysis of α-terpinene, a process that necessitates maintaining reaction temperatures as low as -78°C. This cryogenic requirement not only imposes a massive energy burden on manufacturing facilities but also introduces severe safety risks associated with the handling of unstable ozonide intermediates and the use of dimethyl sulfide for reduction. Furthermore, these traditional oxidative cleavage methods often suffer from poor regioselectivity, yielding complex mixtures where the desired product constitutes merely 30% of the output, accompanied by substantial amounts of isomeric impurities like 3-isopropyl-6-carbonyl-2-heptenal. Subsequent attempts using solid acid catalysts or photo-oxidation techniques have similarly struggled with issues regarding catalyst availability, harsh reaction conditions, and the generation of difficult-to-remove byproducts, rendering them unsuitable for cost-effective industrial scale-up.
The Novel Approach
In stark contrast to these cumbersome legacy protocols, the novel approach outlined in the patent leverages a straightforward acid-catalyzed rearrangement and dehydration sequence that operates efficiently at room temperature. By dissolving the precursor, 4H-5-(1-hydroxy-1-methylethyl)-2-methyl-2-furanacetaldehyde, in a 45-55% aqueous sulfuric acid solution, the reaction proceeds smoothly over a period of 12 to 18 hours without the need for external heating or cooling. This methodological simplification eliminates the capital expenditure associated with specialized cryogenic reactors and ozone generators, directly addressing the cost reduction in flavor and fragrance intermediate manufacturing. The reaction mechanism is inherently cleaner, favoring the formation of the target carbonyl structure through a controlled ring-opening of the furan moiety. Moreover, the workup procedure is remarkably benign, involving simple neutralization with sodium hydroxide and standard solvent extraction, which significantly lowers the barrier to entry for manufacturers seeking to integrate this high-value intermediate into their production lines.
Mechanistic Insights into Acid-Catalyzed Furan Ring Opening
The core of this technological breakthrough lies in the elegant mechanistic pathway that transforms the cyclic furan precursor into the linear enone product. Under the acidic conditions provided by the sulfuric acid medium, the tertiary hydroxyl group on the side chain of the starting material undergoes protonation, facilitating the loss of a water molecule to generate a stabilized carbocation intermediate, designated as species (I) in the reaction scheme. This cationic center acts as the driving force for the subsequent structural reorganization. The electron-rich oxygen within the furan ring assists in stabilizing this charge, eventually leading to the rupture of the cyclic ether bond. This ring-opening event is coupled with a hydration step that generates the intermediate aldehyde-ketone species (II), effectively linearizing the carbon skeleton while preserving the critical oxidation states required for the final product.
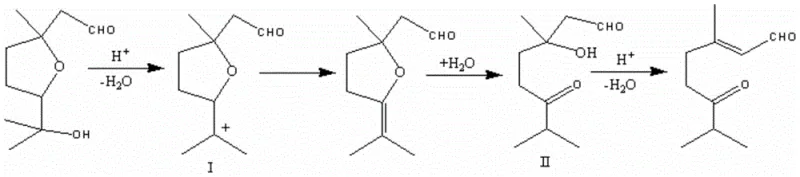
Following the formation of intermediate (II), the system undergoes a final acid-catalyzed dehydration step. The proximity of the hydroxyl group to the newly formed carbonyl functionality allows for the elimination of another water molecule, establishing the conjugated double bond system characteristic of 3,7-dimethyl-6-carbonyl-2-octenal. This cascade of dehydration and rearrangement events is highly specific, minimizing the formation of random polymeric byproducts that often plague acid treatments of sensitive aldehydes. From an impurity control perspective, the use of a homogeneous liquid acid catalyst ensures uniform mixing and heat distribution, preventing localized hot spots that could lead to charring or over-oxidation. The result is a crude reaction mixture with a high assay of the target compound, which simplifies the downstream purification burden and ensures that the final isolated material meets the stringent purity specifications demanded by the fine chemical industry.
How to Synthesize 3,7-Dimethyl-6-Carbonyl-2-Octenal Efficiently
The practical execution of this synthesis is designed for operational simplicity, making it highly attractive for process chemists aiming to transfer laboratory protocols to pilot or production scales. The procedure begins with the careful dissolution of the furan-based starting material into the aqueous sulfuric acid medium, where the concentration of the acid is tightly controlled between 45% and 55% by mass to balance reaction rate and selectivity. Following the reaction period, the mixture is carefully neutralized to prevent acid-catalyzed degradation of the product during workup. The detailed standardized synthesis steps, including specific molar ratios, agitation speeds, and chromatographic gradients, are provided in the structured guide below to ensure reproducibility and safety compliance.
- Dissolve the starting material 4H-5-(1-hydroxy-1-methylethyl)-2-methyl-2-furanacetaldehyde in a 45-55% aqueous sulfuric acid solution and stir at room temperature for 12 to 18 hours to facilitate ring-opening and dehydration.
- Neutralize the reaction mixture to pH 7 using a 4-8% sodium hydrox aqueous solution, then perform liquid-liquid extraction using diethyl ether to isolate the organic phase containing the crude product.
- Dry the organic layer with anhydrous magnesium sulfate, filter, concentrate under reduced pressure, and purify the residue via silica gel column chromatography using a petroleum ether and ethyl acetate gradient.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this acid-catalyzed synthesis offers profound strategic benefits that extend beyond mere technical feasibility. The elimination of cryogenic requirements and hazardous oxidants fundamentally alters the cost structure of producing this intermediate, removing the dependency on specialized infrastructure that often creates bottlenecks in chemical supply chains. By utilizing commodity chemicals like sulfuric acid and sodium hydroxide, the process insulates manufacturers from the price volatility associated with exotic catalysts or rare metal reagents. Furthermore, the ambient temperature operation significantly reduces the facility's energy footprint, aligning with modern sustainability goals and reducing utility costs. The robustness of the reaction also implies a higher success rate per batch, minimizing waste and maximizing the throughput of valuable reactor time, which is a critical metric for maintaining supply continuity in a competitive market.
- Cost Reduction in Manufacturing: The most immediate financial impact stems from the drastic simplification of the reaction conditions. By operating at room temperature, the process completely negates the need for energy-intensive refrigeration systems required by older ozonolysis methods, leading to substantial operational savings. Additionally, the replacement of expensive and difficult-to-handle reagents like ozone and dimethyl sulfide with inexpensive mineral acids results in a significantly lower raw material cost per kilogram of product. The high yield and selectivity of the reaction further contribute to cost efficiency by reducing the volume of waste solvents and the load on purification systems, ensuring that the overall cost of goods sold is optimized for maximum margin retention.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of widely available and stable starting materials. The precursor, 4H-5-(1-hydroxy-1-methylethyl)-2-methyl-2-furanacetaldehyde, is chemically accessible, avoiding the supply constraints often associated with natural terpenes that are subject to agricultural variability. The simplicity of the reagents—sulfuric acid and common organic solvents like diethyl ether—means that production is not vulnerable to shortages of niche catalysts. This reliability allows for more accurate forecasting and inventory planning, ensuring that downstream customers in the flavor and pharmaceutical industries receive consistent deliveries without the risk of production halts due to reagent unavailability.
- Scalability and Environmental Compliance: Scaling this process from laboratory to commercial production is inherently safer and more straightforward due to the absence of explosive ozonides and the use of non-toxic acid catalysts. The aqueous nature of the reaction medium facilitates easier heat management in large reactors, mitigating the risk of thermal runaways. From an environmental standpoint, the process generates less hazardous waste compared to heavy metal-catalyzed or ozonolysis routes, simplifying effluent treatment and disposal compliance. The ability to achieve high purity through standard silica gel chromatography using common eluents further ensures that the final product meets regulatory standards for use in food and medical applications without requiring complex remediation steps.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the viability of this synthesis for their specific applications, we have compiled answers to common inquiries regarding the process parameters and product quality. These insights are derived directly from the experimental data and technical disclosures within the patent documentation, providing a transparent view of the method's capabilities. Understanding these details is crucial for assessing the fit of this intermediate within your existing formulation or synthetic workflows.
Q: What are the primary advantages of this acid-catalyzed method over traditional ozonolysis?
A: Unlike traditional methods requiring cryogenic conditions (-78°C) and expensive ozone generators, this patented process operates efficiently at room temperature using dilute sulfuric acid, drastically reducing energy consumption and equipment complexity while eliminating hazardous ozonide intermediates.
Q: How is high purity (>98%) achieved in the final product?
A: High purity is secured through a robust two-stage purification strategy: initial neutralization and solvent extraction remove bulk acidic impurities, followed by precise silica gel column chromatography using a optimized petroleum ether to ethyl acetate ratio (7:1 to 12:1) to separate the target aldehyde from isomeric byproducts.
Q: Is the starting material 4H-5-(1-hydroxy-1-methylethyl)-2-methyl-2-furanacetaldehyde readily available?
A: Yes, the starting furan derivative is chemically accessible and cost-effective compared to the terpenoid precursors used in older synthetic routes, ensuring a stable supply chain foundation for large-scale manufacturing of this norisoprenoid derivative.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,7-Dimethyl-6-Carbonyl-2-Octenal Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of your final products. Our team of expert process chemists has thoroughly analyzed the technological potential of this acid-catalyzed route and is fully prepared to support its implementation. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop to bulk manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 3,7-dimethyl-6-carbonyl-2-octenal we supply meets the highest international standards for consistency and performance.
We invite you to collaborate with us to leverage this advanced synthesis for your supply chain needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this method can optimize your budget. Please contact us today to request specific COA data and comprehensive route feasibility assessments, and let us help you secure a reliable, cost-effective source for this essential fine chemical intermediate.