Scalable Synthesis of Pseudouridine via Novel Tribenzyloxy-D-Ribono-1,4-Lactone Route for mRNA Therapeutics
The landscape of nucleoside analog manufacturing is undergoing a significant transformation, driven by the surging demand for mRNA therapeutics and the critical need for high-purity modified bases. Patent CN114940676A introduces a groundbreaking methodology for the synthesis of pseudouridine, a vital "fifth base" in RNA that enhances the stability and translational efficiency of mRNA drugs. This technical disclosure moves away from the limitations of traditional enzymatic pathways and unstable chemical routes, proposing a robust three-step synthesis centered on 2,3,5-tribenzyloxy-D-ribono-1,4-lactone. For R&D directors and supply chain leaders, this patent represents a pivotal shift towards a more reliable pharmaceutical intermediate supplier model, offering a pathway that is chemically defined, scalable, and economically viable for industrial production.
The core innovation lies in the strategic selection of starting materials and reaction conditions that mitigate the purification nightmares associated with previous methods. By utilizing a fully benzyl-protected ribose lactone, the process ensures that the sugar moiety remains intact and stereochemically defined throughout the rigorous organometallic transformations. This approach directly addresses the historical bottlenecks of low yield and difficult isolation, positioning this technology as a cornerstone for cost reduction in pharmaceutical intermediates manufacturing. The following analysis dissects the mechanistic advantages and commercial implications of this novel synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of pseudouridine has been plagued by significant technical hurdles that hindered its widespread adoption in large-scale drug manufacturing. Traditional biological enzyme catalysis methods, while specific, suffer from inherently low conversion rates and are heavily dependent on the selectivity of the biocatalyst, often resulting in pseudouridine appearing merely as a minor byproduct that is arduous to separate from the reaction matrix. Furthermore, earlier chemical synthesis strategies, such as those reported by Hanessian et al., relied on acetonide protecting groups which demonstrated poor stability under the requisite reaction conditions. These legacy routes frequently encountered issues with difficult post-reaction purification, leading to suboptimal yields that rendered them unsuitable for the commercial scale-up of complex pharmaceutical intermediates. The instability of protecting groups often led to side reactions, generating impurity profiles that were unacceptable for clinical-grade material.
The Novel Approach
In stark contrast, the methodology disclosed in CN114940676A leverages the robustness of benzyl ether protecting groups to create a streamlined and high-yielding process. The synthesis initiates with the addition of 2,4-dialkoxy-5-bromopyrimidine to 2,3,5-tribenzyloxy-D-ribono-1,4-lactone, followed by a specialized reduction and a final deprotection sequence. This route eliminates the volatility associated with acetonide groups and avoids the biological variability of enzymatic processes. As illustrated in the reaction scheme below, the transformation is direct and efficient, utilizing standard organic reagents that are readily available in the global chemical market.
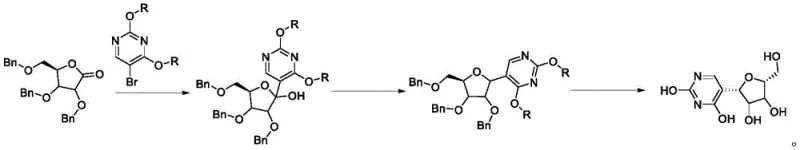
This novel approach not only simplifies the operational workflow but also drastically improves the purity profile of the final product. The use of stable benzyl groups allows for harsher reaction conditions necessary for C-C bond formation without compromising the integrity of the sugar ring. Consequently, this method offers a viable solution for reducing lead time for high-purity pseudouridine production, enabling manufacturers to meet the rigorous quality standards required for mRNA vaccine and therapeutic development.
Mechanistic Insights into Organolithium Addition and Silane Reduction
The success of this synthesis hinges on the precise execution of the initial carbon-carbon bond-forming step. In the first stage, 2,4-dialkoxy-5-bromopyrimidine undergoes lithiation, typically using n-butyllithium at cryogenic temperatures ranging from -70°C to -60°C in tetrahydrofuran (THF). This generates a highly reactive pyrimidyl lithium species which subsequently attacks the carbonyl carbon of the 2,3,5-tribenzyloxy-D-ribono-1,4-lactone. The result is the formation of Intermediate 1, a hemiketal-like structure where the pyrimidine base is attached to the anomeric position. The structure of this critical intermediate, shown below, highlights the retention of the benzyl protecting groups which shield the secondary hydroxyls from unwanted side reactions during this nucleophilic attack.
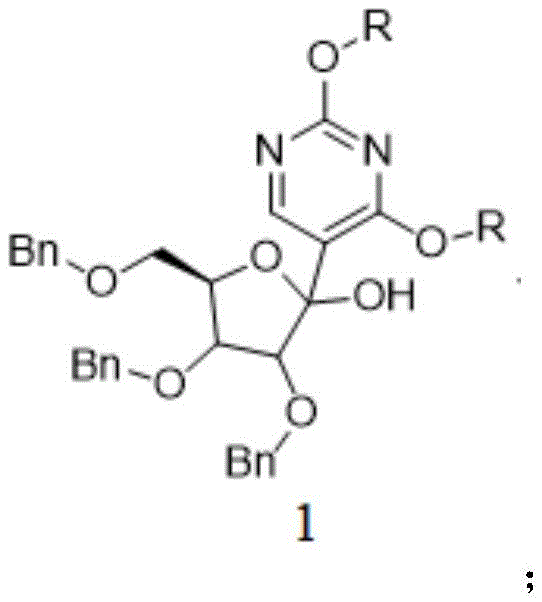
Following the addition, the process employs a sophisticated reduction strategy to establish the stable C-glycosidic bond characteristic of pseudouridine. Intermediate 1 is treated with triethylsilane and boron trifluoride diethyl etherate. This Lewis acid-mediated reduction selectively targets the anomeric hydroxyl group, replacing it with a hydrogen atom to form the methylene linkage found in Intermediate 2. The reaction conditions are meticulously controlled, starting at -70°C and warming to -20°C, to ensure complete conversion while preventing the cleavage of the benzyl ethers. This step is crucial for locking in the beta-configuration of the nucleoside, ensuring the final product possesses the correct stereochemistry required for biological activity.
Impurity control is inherently built into this mechanism through the stability of the intermediates. Unlike acetonide-protected routes where acid sensitivity can lead to ring opening or migration, the benzyl-protected intermediates remain chemically inert to the acidic conditions of the reduction step. This stability minimizes the formation of regioisomers and degradation products, resulting in a cleaner reaction mixture that requires less aggressive purification. The final deprotection using boron trichloride cleanly removes the benzyl groups to reveal the free hydroxyls of pseudouridine, completing the synthesis with high fidelity.
How to Synthesize Pseudouridine Efficiently
The synthesis of pseudouridine via this tribenzyloxy lactone route is designed for operational simplicity and high throughput. The process is divided into three distinct stages: the cryogenic lithiation-addition, the silane-mediated reduction, and the final Lewis acid deprotection. Each step utilizes common industrial solvents and reagents, facilitating easy technology transfer from the laboratory to pilot and production scales. The detailed standardized synthesis steps, including specific molar ratios and quenching protocols, are outlined in the guide below to ensure reproducibility and safety.
- Perform a cryogenic addition reaction between 2,3,5-tribenzyloxy-D-ribono-1,4-lactone and 2,4-dialkoxy-5-bromopyrimidine using n-butyllithium in THF at -70°C to -60°C to form Intermediate 1.
- Execute a reduction reaction on Intermediate 1 using triethylsilane and boron trifluoride diethyl etherate at temperatures ranging from -70°C to -20°C to generate Intermediate 2.
- Conduct a final deprotection reaction on Intermediate 2 using boron trichloride in dichloromethane, warming from -70°C to room temperature to yield crude pseudouridine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this chemical synthesis route offers profound strategic benefits beyond mere technical feasibility. The reliance on stable, commercially available starting materials like 2,3,5-tribenzyloxy-D-ribono-1,4-lactone and simple pyrimidine derivatives decouples production from the supply constraints often associated with specialized enzymes or unstable precursors. This shift ensures a more predictable supply chain, reducing the risk of production stoppages due to raw material shortages. Furthermore, the elimination of complex biological fermentation and downstream processing steps significantly streamlines the manufacturing timeline.
- Cost Reduction in Manufacturing: The proposed synthetic route achieves substantial cost savings by eliminating the need for expensive biocatalysts and the associated buffer systems required for enzymatic reactions. The chemical reagents used, such as n-butyllithium and triethylsilane, are commodity chemicals available from multiple global suppliers, fostering competitive pricing and reducing dependency on single-source vendors. Additionally, the high stability of the benzyl-protected intermediates reduces material loss during purification, leading to improved overall mass balance and lower cost of goods sold (COGS) per kilogram of active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: By adopting a fully synthetic pathway, manufacturers can achieve greater consistency in batch-to-batch quality, a critical factor for regulatory compliance in the pharmaceutical sector. The robust nature of the reaction conditions means that production is less susceptible to environmental variables that often plague biological processes. This reliability translates to shorter lead times and the ability to rapidly scale up production volumes in response to market demand, ensuring a continuous supply of this critical mRNA component without the delays typical of bio-based manufacturing.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard organic solvents like THF and dichloromethane which can be efficiently recovered and recycled in a closed-loop system. The absence of heavy metal catalysts or toxic biological waste streams simplifies waste management and aligns with increasingly stringent environmental regulations. The straightforward workup procedures, involving standard aqueous quenches and extractions, facilitate the design of continuous flow reactors or large batch vessels, making the transition from grams to tons a seamless engineering challenge rather than a scientific hurdle.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this pseudouridine synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the operational parameters and strategic value of this novel route for industry stakeholders.
Q: Why is the tribenzyloxy protecting group strategy superior for pseudouridine synthesis?
A: The use of 2,3,5-tribenzyloxy-D-ribono-1,4-lactone provides exceptional chemical stability compared to acetonide protecting groups used in prior art. This stability prevents premature degradation during the harsh lithiation and reduction steps, ensuring higher overall purity and simplifying the downstream purification process for pharmaceutical applications.
Q: How does this chemical route address the scalability issues of enzymatic methods?
A: Unlike enzymatic catalysis which suffers from low conversion rates and difficult separation of biocatalysts, this purely chemical route utilizes standard organic reagents like n-butyllithium and triethylsilane. This allows for precise control over reaction kinetics and straightforward workup procedures, making it inherently suitable for multi-kilogram to ton-scale manufacturing without biological variability.
Q: What are the critical temperature controls required for the reduction step?
A: The reduction of Intermediate 1 requires strict thermal management, initiating at -70°C to -65°C during triethylsilane addition and gradually warming to -30°C to -20°C upon introduction of boron trifluoride. This gradient is essential to control the exotherm and ensure selective reduction of the anomeric hydroxyl group without affecting the benzyl protecting groups.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pseudouridine Supplier
As the demand for mRNA therapeutics continues to accelerate, securing a stable supply of high-quality nucleoside modifications like pseudouridine is paramount for drug developers. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, leveraging advanced synthetic methodologies such as the tribenzyloxy lactone route to deliver superior products. Our facility boasts extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric needs of global pharmaceutical partners. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the exacting standards required for clinical and commercial applications.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in complex nucleoside synthesis can drive value for your organization.