Advanced Synthesis of Pyrimidinedione Intermediates for Commercial DPP-IV Inhibitor Production
Introduction to Next-Generation DPP-IV Inhibitor Intermediates
The global demand for effective Type 2 diabetes treatments continues to drive innovation in the synthesis of Dipeptidyl Peptidase-IV (DPP-IV) inhibitors. A pivotal development in this sector is detailed in patent CN103030631A, which introduces a novel intermediate compound essential for producing pyrimidinedione-class DPP-IV inhibitors. This technology addresses critical bottlenecks in the manufacturing of drugs such as 2-({6-[(3R)-3-aminopiperidine-1-yl]-3-methyl-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl}methyl)benzonitrile. As a reliable pharmaceutical intermediate supplier, understanding the structural nuances and synthetic efficiency of these molecules is paramount for ensuring a stable supply chain for downstream API manufacturers. The structural complexity of these inhibitors requires precise stereochemical control, particularly at the piperidine ring, to maintain biological efficacy.
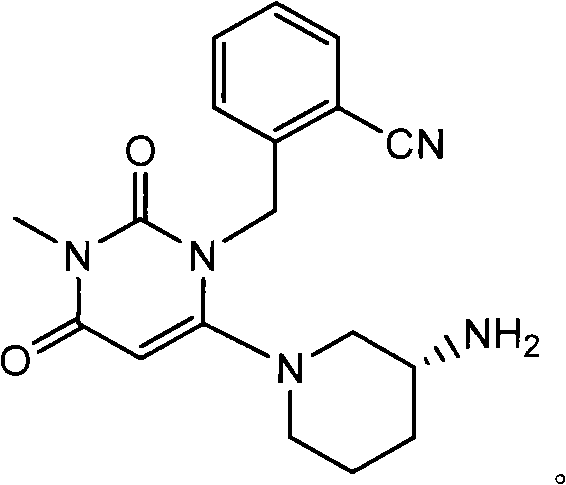
The invention specifically targets the optimization of the synthetic pathway to overcome the limitations of earlier methods which suffered from low yields and difficult purification processes. By introducing a protected intermediate strategy, the patent outlines a method that not only enhances the chemical yield but also simplifies the isolation of the final product. This advancement represents a significant leap forward in cost reduction in API manufacturing, as it reduces the consumption of raw materials and minimizes waste generation. For procurement managers and supply chain heads, this translates to a more predictable and economically viable sourcing option for high-value diabetes medication precursors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the innovations disclosed in CN103030631A, the synthesis of these critical intermediates relied on a linear alkylation sequence that was fraught with inefficiencies. As illustrated in the prior art reaction scheme, the conventional route involved the direct alkylation of 6-chlorouracil followed by methylation and subsequent coupling with the aminopiperidine moiety. This traditional approach resulted in a cumulative yield of merely approximately 20%, which is commercially unsustainable for large-scale production. Furthermore, the crude product obtained from this method contained significant impurities that necessitated rigorous and costly purification steps, such as column chromatography, which are notoriously difficult to scale up in an industrial setting.
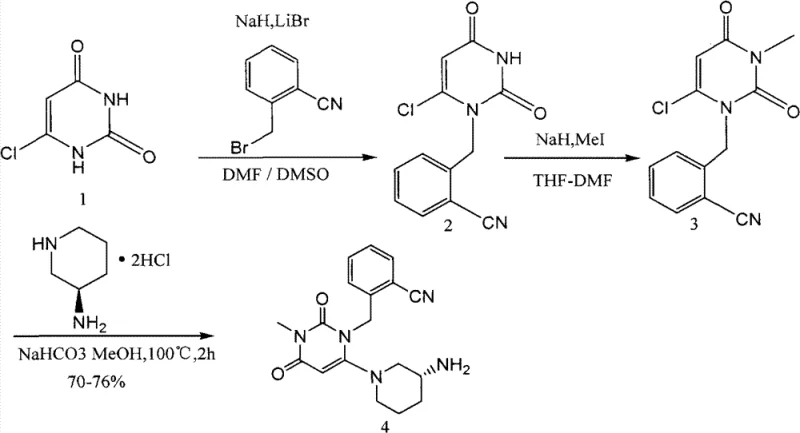
The reliance on mixed solvent systems like DMF-DMSO for the initial alkylation often led to side reactions and the formation of by-products that were chemically similar to the target molecule, making separation arduous. The low purity of the intermediate compounded the challenges in the final drug substance manufacturing, potentially affecting the safety profile and regulatory approval of the final API. For a commercial scale-up of complex pharmaceutical intermediates, such low efficiency and high impurity loads are unacceptable barriers that increase the lead time and overall cost of goods sold.
The Novel Approach
The novel approach presented in the patent fundamentally restructures the synthetic logic by employing a "protect-then-couple" strategy. Instead of attempting to couple the free amine directly under harsh conditions, the synthesis begins with the protection of the chiral piperidine amine using a phthalimide group. This creates a robust intermediate (Compound 1-1) that is stable under the subsequent alkylation conditions. The key coupling reaction then proceeds between this protected piperidine and the chlorouracil derivative (Compound 1-2) in the presence of N-Methyl pyrrolidone (NMP) and diisopropylethylamine (DIPEA). This modification allows the reaction to proceed at elevated temperatures (120-130 °C) with exceptional efficiency, achieving yields as high as 90-93.6% for the protected intermediate.
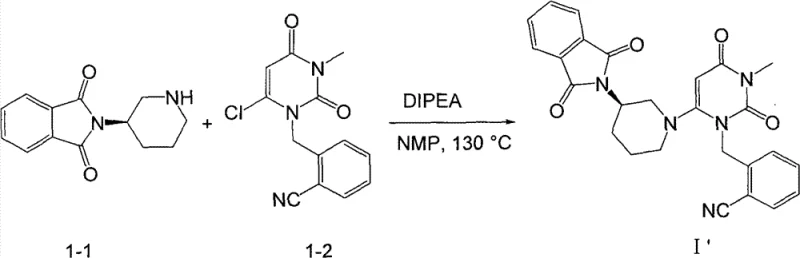
Following the coupling, the phthalimide protecting group is cleanly removed using hydrazine hydrate under reflux conditions in ethanol. This deprotection step is highly selective and proceeds with a remarkable yield of 95%, delivering the final free amine product with high purity. This two-step sequence (coupling followed by deprotection) effectively bypasses the impurity issues of the old route and eliminates the need for chromatographic purification, relying instead on simple crystallization techniques. This shift represents a paradigm change in how we approach the synthesis of high-purity pharmaceutical intermediates, prioritizing atom economy and operational simplicity.
Mechanistic Insights into Nucleophilic Substitution and Deprotection
The core of this synthetic breakthrough lies in the mechanistic efficiency of the nucleophilic substitution reaction facilitated by the phthalimide protecting group. In the coupling step, the nitrogen atom of the protected piperidine acts as a nucleophile, attacking the C-6 position of the chlorouracil ring where the chlorine atom serves as a leaving group. The use of NMP as a polar aprotic solvent is critical here, as it stabilizes the transition state and enhances the nucleophilicity of the amine without solvating it too strongly, which would hinder reactivity. The addition of DIPEA serves as a non-nucleophilic base to scavenge the hydrochloric acid generated during the reaction, driving the equilibrium towards product formation. The thermal energy provided at 120-130 °C ensures that the activation energy barrier is overcome rapidly, leading to complete conversion of the starting materials.
The subsequent deprotection mechanism utilizes the high affinity of hydrazine for the carbonyl groups of the phthalimide ring. Hydrazine hydrate attacks the imide carbonyls, leading to the formation of phthalhydrazide and the release of the free primary amine on the piperidine ring. This reaction is thermodynamically favorable and proceeds cleanly in ethanol, a green and easily recoverable solvent. The mechanistic elegance of this route ensures that the chiral center at the 3-position of the piperidine ring remains intact throughout the process, preserving the stereochemical integrity required for the biological activity of the final DPP-IV inhibitor. This level of control is essential for meeting the stringent regulatory requirements for chiral drugs.
Impurity control is inherently built into this mechanism. By masking the reactive amine during the harsh alkylation phase, the formation of bis-alkylated by-products or polymeric impurities is effectively suppressed. The resulting intermediate (Compound I') is a solid that can be easily isolated by filtration and washing, removing soluble impurities before the final deprotection. This sequential purification strategy ensures that the final product meets the rigorous purity specifications demanded by the pharmaceutical industry, typically requiring single impurities to be below 0.1% and total impurities below 1.0%.
How to Synthesize Pyrimidinedione Intermediate Efficiently
The synthesis of this high-value intermediate is designed for operational robustness, making it ideal for transfer from laboratory to pilot and commercial plant scales. The process begins with the preparation of the protected amine, followed by the critical coupling reaction in NMP, and concludes with the hydrazine-mediated deprotection. Each step has been optimized to maximize yield and minimize solvent usage, aligning with modern green chemistry principles. The detailed standardized operating procedures for this synthesis, including specific temperature ramps, addition rates, and work-up protocols, are critical for reproducibility.
- Synthesize the protected piperidine intermediate (Compound 1-1) by reacting R-3-aminopiperidine dihydrochloride with phthalic anhydride in acetic acid at 130 °C.
- Perform the key coupling reaction between Compound 1-1 and the chlorouracil derivative (Compound 1-2) using NMP and DIPEA at 120-130 °C to form the protected intermediate I'.
- Execute the deprotection step by refluxing intermediate I' with hydrazine hydrate in ethanol to yield the final free amine product with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers transformative benefits that extend beyond simple chemistry. The primary advantage is the drastic improvement in process efficiency, which directly correlates to lower manufacturing costs and enhanced supply security. By moving away from low-yielding, chromatography-dependent methods to a high-yield crystallization-based process, manufacturers can significantly reduce the cost of goods sold (COGS). This efficiency gain allows for more competitive pricing strategies in the global market for diabetes medications, providing a distinct advantage to companies that secure access to this technology.
- Cost Reduction in Manufacturing: The elimination of column chromatography is a major cost driver reduction. Chromatography is expensive due to the high cost of silica gel, large solvent volumes, and low throughput. By replacing this with crystallization, the process becomes vastly more economical. Furthermore, the increase in yield from ~20% to over 90% in key steps means that nearly five times less raw material is required to produce the same amount of product. This substantial reduction in material consumption directly lowers the variable costs associated with production, allowing for better margin management and price stability for long-term contracts.
- Enhanced Supply Chain Reliability: The starting materials for this route, such as R-3-aminopiperidine dihydrochloride, phthalic anhydride, and 6-chloro-3-methyluracil, are commodity chemicals with established global supply chains. This reduces the risk of supply disruptions compared to routes requiring exotic or custom-synthesized reagents. Additionally, the robustness of the reaction conditions (using standard solvents like NMP and ethanol) means that the process can be executed in a wide range of multipurpose chemical plants without requiring specialized equipment. This flexibility ensures reducing lead time for high-purity pharmaceutical intermediates and guarantees a continuous flow of material to downstream API manufacturers.
- Scalability and Environmental Compliance: The process is inherently scalable. The use of heterogeneous reactions that result in solid products facilitates easy filtration and handling on a multi-ton scale. Moreover, the solvent system is relatively benign; ethanol and water are used in the final steps, reducing the environmental footprint compared to processes relying heavily on chlorinated solvents or toxic amides throughout. The ability to recycle solvents like NMP and dichloromethane further enhances the sustainability profile of the manufacturing process. This alignment with environmental, social, and governance (ESG) goals is increasingly important for multinational pharmaceutical companies when selecting suppliers.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this pyrimidinedione intermediate. These answers are derived directly from the technical specifications and experimental data provided in the patent literature, ensuring accuracy and relevance for technical decision-makers.
Q: What is the primary advantage of the new synthesis route over conventional methods?
A: The new route described in CN103030631A utilizes a phthalimide protecting group strategy which significantly improves the overall yield from approximately 20% in prior art to over 90% in the coupling step, while eliminating the need for complex column chromatography purification.
Q: How is the final purity of the intermediate ensured without chromatography?
A: The process relies on robust crystallization techniques. Specifically, the final benzoate salt can be recrystallized from an ethanol and ethyl acetate mixed solvent system, achieving purity levels exceeding 99.6% suitable for pharmaceutical applications.
Q: Is this process scalable for industrial manufacturing?
A: Yes, the use of common solvents like NMP, ethanol, and dichloromethane, combined with high-yield reactions that do not require sensitive catalysts or extreme pressures, makes this route highly amenable to commercial scale-up from kilogram to multi-ton production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Pyrimidinedione Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving diabetes medications. Our technical team has thoroughly analyzed the synthetic pathways described in CN103030631A and is fully equipped to implement this advanced chemistry at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, including chiral purity and residual solvent analysis, to guarantee that every batch meets international pharmacopeial standards.
We invite you to collaborate with us to leverage this superior synthetic route for your DPP-IV inhibitor projects. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data, route feasibility assessments, and sample quantities. Let us help you optimize your supply chain and accelerate your time to market with our reliable and cost-effective manufacturing solutions.