Advanced Synthesis of Iodixanol Impurities for Quality Control and Commercial Scale-up
Advanced Synthesis of Iodixanol Impurities for Quality Control and Commercial Scale-up
Introduction to Patent CN110590591B and Contrast Agent Quality
The pharmaceutical industry relies heavily on the precise characterization of non-ionic iodinated contrast agents like iodixanol and iohexol to ensure patient safety and diagnostic accuracy. Patent CN110590591B introduces a groundbreaking preparation method for specific impurities that naturally arise during the manufacturing of these critical diagnostic tools. These impurities, particularly 5-[N-(2-hydroxypropyl acetate) acetamido]-2,4,6-triiodo-N,N'-bis(2,3-dihydroxypropyl)-1,3-benzenedicarboxamide, possess polarity profiles nearly identical to the active pharmaceutical ingredient, making them notoriously difficult to separate and quantify using standard analytical techniques. The absence of a reliable synthesis route for these reference standards in prior art has long hindered the ability of quality control laboratories to establish rigorous purity specifications. By providing a robust and reproducible method to generate these specific molecular structures, this technology empowers manufacturers to implement stricter quality assurance protocols, ultimately reducing the risk of adverse reactions and ensuring compliance with global regulatory standards for injectable contrast media.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the identification and quantification of process-related impurities in iodixanol synthesis have been plagued by significant technical challenges due to the lack of authentic reference standards. In conventional manufacturing scenarios, these impurities are generated inadvertently through the hydrolysis of intermediate compounds, leading to complex mixtures where the target impurity co-elutes with the main product during chromatographic analysis. This overlap creates a blind spot in quality control, forcing manufacturers to rely on indirect estimation methods that lack precision and reliability. Furthermore, the structural similarity between the impurity and the final drug substance means that standard purification techniques often fail to isolate the contaminant in sufficient quantities for structural elucidation or toxicity testing. This limitation not only compromises the integrity of the batch release data but also poses a potential regulatory risk, as health authorities increasingly demand comprehensive impurity profiling for all new drug applications and generic filings.
The Novel Approach
The innovative strategy outlined in the patent data overcomes these historical barriers by establishing a dedicated synthetic pathway specifically designed to produce the target impurity as a standalone chemical entity. Instead of relying on random formation during the main production run, this method utilizes a controlled two-step reaction sequence that selectively constructs the impurity's unique molecular architecture. By starting with 5-acetamido-2,4,6-triiodo-N,N-bis-(2,3-dihydroxypropyl)-1,3-benzenedicarboxamide and reacting it with 2-hydroxy-3-chloropropyl acetate under optimized conditions, the process ensures high conversion rates while minimizing the formation of secondary byproducts. This deliberate synthesis allows for the production of gram-to-kilogram quantities of the impurity standard, enabling laboratories to calibrate their analytical instruments with high confidence. The result is a dramatic improvement in the resolution of chromatographic peaks, allowing for the precise detection of trace contaminants that were previously invisible to standard quality control measures.
Mechanistic Insights into FeCl3-Catalyzed Esterification and Substitution
The core of this synthesis lies in a sophisticated two-stage chemical transformation that leverages specific catalytic and environmental controls to drive reaction efficiency. The first stage involves the preparation of 2-hydroxy-3-chloropropyl acetate through the reaction of epichlorohydrin and acetic acid, catalyzed by anhydrous ferric chloride. This Lewis acid catalyst plays a crucial role in activating the epoxide ring, facilitating the nucleophilic attack by the acetic acid while maintaining a reaction temperature that prevents polymerization or degradation of the sensitive halogenated intermediate. The subsequent step involves the nucleophilic substitution of the chloro-group on the acetate with the amine functionality of the triiodobenzene derivative. This reaction is highly sensitive to pH levels, requiring a carefully maintained alkaline environment between pH 11.5 and 12 to ensure the deprotonation of the amine without causing hydrolysis of the ester or amide bonds. 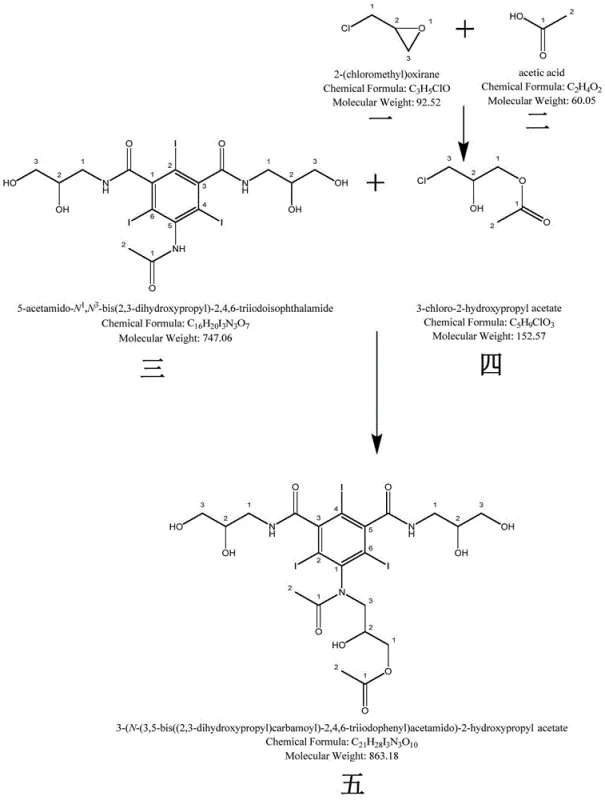
Understanding the structural nuances of the final impurity is essential for R&D teams aiming to replicate this synthesis or develop analogous purification methods. The molecule features a complex triiodinated benzene core substituted with multiple hydroxypropyl and acetamido groups, creating a highly polar and sterically hindered structure. ![Chemical structure of 5-[N-(2-hydroxypropyl acetate) acetamido]-2,4,6-triiodo-N,N'-bis(2,3-dihydroxypropyl)-1,3-benzenedicarboxamide](/insights/img/iodixanol-impurity-synthesis-pharma-supplier-20260314231446-01.webp)
The presence of three iodine atoms significantly increases the molecular weight and X-ray absorption properties, which is critical for its function as a contrast agent impurity but also complicates its purification. The specific arrangement of the hydroxyl and acetate groups dictates the molecule's solubility profile, making it soluble in polar aprotic solvents like dimethylacetamide but requiring aqueous workups for isolation. The patent highlights that controlling the pH during the termination of the reaction is vital; adjusting to a neutral pH of 6 prevents the formation of gelatinous precipitates that can trap the product and reduce recovery yields. This mechanistic understanding allows process chemists to fine-tune reaction parameters, such as dropwise addition rates and stirring speeds, to maximize the formation of the desired isomer while suppressing the generation of regioisomers that could further complicate the impurity profile.
How to Synthesize Iodixanol Impurity Efficiently
Implementing this synthesis route requires strict adherence to the operational parameters defined in the patent to ensure reproducibility and safety on a commercial scale. The process begins with the careful handling of epichlorohydrin and acetic acid, where temperature control is paramount to manage the exothermic nature of the esterification reaction. Following the isolation of the chloro-acetate intermediate, the second reaction step demands precise pH monitoring using bases like sodium methoxide or sodium hydroxide to maintain the optimal alkalinity window. The reaction mixture must be stirred for an extended period, typically around 16 hours, to ensure complete conversion of the starting materials before proceeding to the purification phase. Detailed standardized synthesis steps see the guide below.
- Prepare 2-hydroxy-3-chloropropyl acetate by reacting epichlorohydrin with acetic acid using anhydrous ferric chloride as a catalyst under controlled temperature conditions.
- React 5-acetamido-2,4,6-triiodo-N,N-bis-(2,3-dihydroxypropyl)-1,3-benzenedicarboxamide with the prepared acetate in a solvent like DMA, adjusting pH to 11.5-12.
- Purify the crude reaction mixture using macroporous resin such as XAD-1600N to isolate the high-purity impurity standard.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method offers tangible benefits that extend beyond mere technical compliance, directly impacting the bottom line and operational resilience. By establishing an in-house or partnered capability to produce critical impurity standards, companies can significantly reduce their reliance on external suppliers who often charge premium prices for small quantities of reference materials. This self-sufficiency mitigates the risk of supply disruptions that can halt quality control testing and delay batch releases, ensuring a smoother flow of finished goods to the market. Furthermore, the use of macroporous resin purification instead of traditional preparative HPLC drastically lowers the cost of goods sold by reducing solvent consumption and increasing throughput capacity. The scalability of this process means that as production volumes of iodixanol increase, the supply of necessary quality control standards can be scaled proportionally without incurring exponential cost increases.
- Cost Reduction in Manufacturing: The elimination of expensive chromatographic purification steps in favor of macroporous resin adsorption represents a significant operational cost saving. Traditional methods for isolating polar impurities often require large volumes of high-grade organic solvents and specialized preparative columns, which are costly to purchase, maintain, and dispose of. By utilizing robust industrial resins like XAD-1600N, the process simplifies the downstream processing workflow, reducing both material costs and labor hours associated with complex fraction collection. Additionally, the high conversion rates achieved through optimized pH and temperature control minimize the waste of valuable iodinated starting materials, which are among the most expensive components in the synthesis of contrast agents. This efficiency translates directly into a lower cost per gram of the impurity standard, allowing quality control budgets to be allocated more effectively across other critical testing areas.
- Enhanced Supply Chain Reliability: Securing a reliable source of impurity standards is crucial for maintaining uninterrupted manufacturing operations, especially in the highly regulated pharmaceutical sector where batch release depends on passing strict purity tests. This synthesis route utilizes readily available raw materials such as epichlorohydrin, acetic acid, and common solvents like dimethylacetamide, which are widely produced and less susceptible to geopolitical supply shocks compared to specialized reagents. The robustness of the reaction conditions, which tolerate slight variations in temperature and pH without significant yield loss, further enhances supply chain stability by reducing the rate of batch failures. By integrating this synthesis into the broader supply chain strategy, companies can build a buffer stock of critical reference materials, insulating themselves from market volatility and ensuring that quality assurance protocols never become a bottleneck for production schedules.
- Scalability and Environmental Compliance: The process is designed with commercial scale-up in mind, utilizing unit operations that are easily transferable from the laboratory to the pilot plant and full-scale production facilities. The use of macroporous resin columns is a well-established technology in the fine chemical industry, allowing for continuous or semi-continuous processing that can handle large volumes of reaction mixture efficiently. From an environmental perspective, the method reduces the generation of hazardous waste by minimizing solvent usage and enabling the recovery and reuse of eluents through membrane concentration techniques. The ability to adjust the pH using common acids and bases simplifies waste treatment processes, ensuring that effluent streams meet regulatory discharge limits without requiring complex neutralization protocols. This alignment with green chemistry principles not only reduces environmental liability but also enhances the company's sustainability profile, which is increasingly important for maintaining partnerships with global pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this impurity synthesis method, providing clarity for stakeholders evaluating its adoption. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, ensuring accuracy and relevance for decision-makers. Understanding these details is essential for assessing the feasibility of integrating this technology into existing quality control workflows and supply chain operations. The information provided here serves as a foundational guide for further discussions with technical teams and potential manufacturing partners.
Q: Why is synthesizing specific impurities necessary for iodixanol production?
A: Impurities generated during iodixanol synthesis have polarities very close to the final product, making them difficult to separate. Having a synthesized standard allows for accurate identification and quantification during quality control.
Q: What purification method is recommended for this impurity?
A: The patent recommends using macroporous resins like XAD-1600N or LX-16. This method effectively separates the impurity from the reaction mixture, achieving purity levels above 95%.
Q: How does the new method improve upon prior art?
A: Prior art lacked a specific preparation method for this impurity. This novel approach provides a controlled synthesis route with optimized pH and temperature conditions to minimize byproducts and ensure high conversion rates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Iodixanol Impurity Supplier
NINGBO INNO PHARMCHEM stands at the forefront of fine chemical manufacturing, offering unparalleled expertise in the scale-up and production of complex pharmaceutical intermediates like the iodixanol impurity described in patent CN110590591B. Our state-of-the-art facilities are equipped to handle the specific requirements of iodinated chemistry, ensuring that every batch meets stringent purity specifications through our rigorous QC labs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, allowing us to seamlessly transition this synthesis from development to full-scale manufacturing. Our commitment to quality ensures that the impurity standards we provide are fully characterized and suitable for use in validated analytical methods, giving our partners confidence in their regulatory submissions.
We invite procurement leaders and technical directors to collaborate with us to optimize their supply chain for contrast agent production. By leveraging our technical capabilities, you can secure a stable supply of high-purity impurity standards while achieving significant cost efficiencies. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing solutions can support your quality goals and business growth.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →