Scalable Synthesis of 3-Fluoro-Oxetane Sulfonate Intermediates for Pharmaceutical Manufacturing
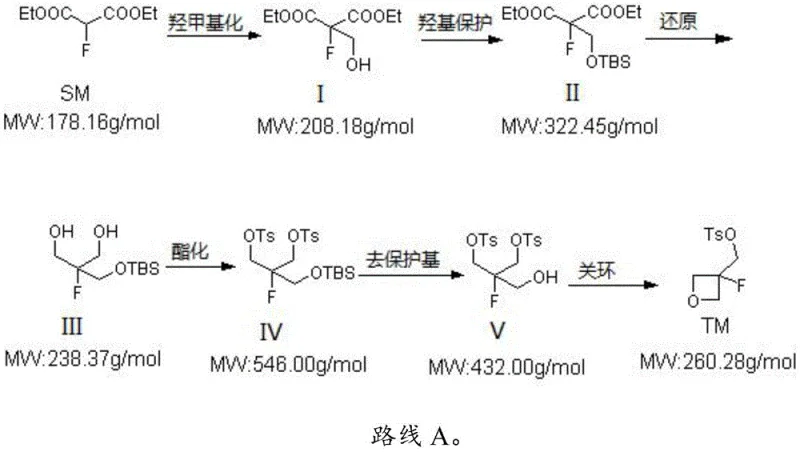
The pharmaceutical industry continuously seeks robust and scalable pathways for constructing complex fluorinated heterocycles, which serve as critical building blocks in modern drug discovery. Patent CN113461643A introduces a significant advancement in this domain by detailing a novel synthesis method for 4-methylbenzenesulfonic acid [(3-fluoro-oxetan-3-yl) methyl] ester. This compound acts as a pivotal precursor for grafting 3-fluoro oxetane fragments into larger molecular architectures, a modification often employed to enhance metabolic stability and bioavailability in active pharmaceutical ingredients. The disclosed methodology utilizes 2-fluoro diethyl malonate as a commercially accessible starting material, navigating through a meticulously optimized six-step sequence that includes hydroxymethylation, hydroxyl protection, reduction, esterification, protecting group removal, and final ring closure. This approach addresses longstanding challenges in the field, offering a pathway that balances high chemical efficiency with operational simplicity, making it particularly attractive for reliable pharmaceutical intermediate supplier networks aiming to secure stable supply chains for next-generation therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3-fluoro-oxetane derivatives has been plagued by significant technical hurdles that impede large-scale manufacturing. Traditional routes often involve the preparation of 3-fluorooxetane-3-methanol followed by tosylation, a process frequently characterized by low total yields and the necessity for labor-intensive column chromatography purification. Such reliance on chromatographic separation is economically prohibitive for kilogram-level amplification, rendering these methods suitable only for small-scale laboratory preparations rather than industrial production. Furthermore, alternative strategies reported in prior art, such as those utilizing benzyl oxymethyl adding followed by debenzylation, introduce severe safety and environmental liabilities. These legacy processes typically demand the use of large quantities of sodium hydride, a highly reactive and dangerous reagent that poses significant explosion risks upon scale-up, alongside the requirement for virulent and expensive boron tribromide for debenzylation steps. These factors collectively create substantial bottlenecks in cost reduction in pharmaceutical intermediate manufacturing, limiting the availability of high-purity intermediates for downstream drug development.
The Novel Approach
In stark contrast to these hazardous and inefficient legacy methods, the novel approach outlined in the patent data presents a streamlined and safer alternative that is inherently designed for commercial scale-up of complex pharmaceutical intermediates. By initiating the synthesis with 2-fluoro diethyl malonate, the route leverages a stable and inexpensive feedstock to construct the carbon skeleton efficiently. The strategy employs a tert-butyldimethylsilyl (TBS) protection group, which offers superior stability and ease of manipulation compared to benzyl groups, thereby eliminating the need for dangerous hydrogenolysis or Lewis acid-mediated cleavage conditions. The subsequent reduction and esterification steps are conducted under mild conditions using common reagents like sodium borohydride and p-toluenesulfonyl chloride, ensuring that the process remains manageable within standard reactor setups. This fundamental redesign of the synthetic logic not only enhances the safety profile of the operation but also simplifies the isolation of intermediates, allowing for direct progression to subsequent steps without rigorous purification, thus driving substantial cost savings and improving the overall throughput of the production line.
Mechanistic Insights into the Six-Step Synthetic Sequence
The core of this technological breakthrough lies in the precise orchestration of functional group transformations that preserve the integrity of the fluorine atom while constructing the strained oxetane ring. The process begins with a hydroxymethylation reaction where 2-fluoro diethyl malonate reacts with formaldehyde in the presence of an inorganic base, effectively installing the necessary hydroxymethyl handle at the alpha position. Following this, the hydroxyl group is protected using tert-butyldimethylsilyl chloride and an organic base, a critical step that prevents unwanted side reactions during the subsequent reduction phase. The reduction of the diester moiety to a diol is achieved using hydride reducing agents, carefully controlled to ensure complete conversion without compromising the silyl ether. The resulting triol intermediate is then selectively esterified at the primary alcohol positions using p-toluenesulfonyl chloride, setting the stage for the final cyclization. The removal of the TBS protecting group reveals the requisite hydroxyl functionality, which then participates in an intramolecular nucleophilic substitution mediated by an organolithium compound. This final ring-closing step is the mechanistic climax, forming the four-membered oxetane ring with high fidelity and establishing the final sulfonate ester structure essential for downstream coupling reactions.
Furthermore, the impurity control mechanism embedded within this route is robust, largely due to the avoidance of harsh conditions that typically generate complex byproduct profiles. The use of mild temperatures, ranging generally from 10°C to 60°C across the various steps, minimizes thermal degradation and polymerization risks that are common in high-energy processes. The selection of solvents such as tetrahydrofuran, ethyl acetate, and methanol ensures good solubility of intermediates while facilitating easy removal during workup. Crucially, the final product is obtained as an off-white solid with an HPLC purity of more than 97%, indicating that the crystallization or pulping steps effectively reject minor impurities generated during the synthesis. This high level of purity is achieved without the need for preparative HPLC or silica gel chromatography, relying instead on standard aqueous workups and solvent exchanges. For R&D directors, this implies a predictable impurity spectrum that is easier to characterize and control, thereby accelerating the regulatory filing process for any drug substance incorporating this key intermediate.
How to Synthesize 4-methylbenzenesulfonic acid [(3-fluoro-oxetan-3-yl) methyl] ester Efficiently
Executing this synthesis requires careful attention to stoichiometry and temperature control, particularly during the exothermic hydroxymethylation and reduction stages. The process is designed to be telescoped where possible, minimizing the hold times of unstable intermediates and maximizing the overall equipment effectiveness. Operators should ensure that the formaldehyde source is of high quality, preferably a 36-38% aqueous solution, to drive the initial condensation to completion. The protection step benefits from the addition of the silyl chloride in batches to manage heat evolution, while the reduction phase requires slow addition of the hydride source to prevent gas evolution issues. Detailed standardized synthesis steps see the guide below, which outlines the specific molar ratios and reaction times optimized in the patent examples to achieve the reported 40-60% total yield. Adhering to these parameters ensures that the process remains within the design space validated for industrial production, guaranteeing consistent quality and batch-to-batch reproducibility.
- Perform hydroxymethylation of 2-fluoro diethyl malonate with formaldehyde and inorganic base to obtain the hydroxymethyl intermediate.
- Protect the hydroxyl group using tert-butyldimethylsilyl chloride and organic base, followed by reduction of the ester groups to alcohols.
- Execute esterification with p-toluenesulfonyl chloride, remove the silyl protecting group, and finalize with organolithium-mediated ring closure.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this synthetic route offers transformative advantages that directly address the pain points of sourcing complex fluorinated building blocks. The elimination of hazardous reagents like sodium hydride and boron tribromide removes significant logistical and disposal costs associated with dangerous goods transportation and waste management. This shift not only lowers the direct cost of goods sold but also mitigates the risk of production stoppages due to safety incidents or regulatory scrutiny regarding hazardous waste. Moreover, the reliance on commodity chemicals such as formaldehyde, sodium bicarbonate, and sodium borohydride ensures that the supply chain is resilient against fluctuations in the availability of exotic or specialized reagents. This stability is crucial for maintaining continuous production schedules and meeting the just-in-time delivery requirements of global pharmaceutical clients who depend on reliable API intermediate supplier partnerships to keep their own pipelines moving.
- Cost Reduction in Manufacturing: The economic viability of this process is significantly enhanced by the removal of column chromatography, a unit operation that is notoriously expensive and difficult to scale. By replacing chromatographic purification with standard extraction, washing, and crystallization techniques, the manufacturing footprint is reduced, and solvent consumption is minimized. The use of inexpensive starting materials like 2-fluoro diethyl malonate further drives down the raw material costs, while the high atom economy of the hydroxymethylation step ensures efficient utilization of feedstocks. These factors combine to create a lean manufacturing process that delivers substantial cost savings without compromising on the quality or purity of the final product, making it an ideal candidate for cost-sensitive generic drug programs.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes directly to improved supply chain reliability and reduced lead time for high-purity intermediates. Because the reactions proceed under mild temperatures and do not require specialized high-pressure or cryogenic equipment, they can be performed in a wide range of multipurpose chemical plants. This flexibility allows for diversified manufacturing locations, reducing the geopolitical and logistical risks associated with single-source suppliers. Additionally, the stability of the intermediates allows for potential stocking strategies, where key precursors can be produced in bulk and held in inventory to buffer against sudden spikes in demand, ensuring that customers receive their orders on time even during periods of market volatility.
- Scalability and Environmental Compliance: Scalability is inherent in the design of this route, as evidenced by the successful demonstration of the process in multi-kilogram batches within the patent examples. The absence of heavy metal catalysts and the use of relatively benign solvents align with green chemistry principles, simplifying the environmental permitting process for new production lines. Waste streams are easier to treat, primarily consisting of aqueous salts and organic solvents that can be recovered and recycled, thereby lowering the environmental impact and ensuring compliance with increasingly stringent global environmental regulations. This forward-thinking approach to process design future-proofs the supply of this intermediate against evolving sustainability mandates in the chemical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this fluorinated oxetane intermediate. These answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for potential partners. Understanding these details is essential for evaluating the feasibility of integrating this intermediate into your specific drug synthesis campaigns and for assessing the long-term viability of the supply partnership.
Q: What are the safety advantages of this new synthesis route compared to conventional methods?
A: This method eliminates the use of hazardous sodium hydride and toxic boron tribromide required in traditional benzyl protection routes, significantly improving process safety and operational feasibility for industrial scale-up.
Q: What is the expected purity and yield of the final 3-fluoro-oxetane sulfonate product?
A: According to patent data, the six-step reaction sequence achieves a total yield ranging from 40% to 60%, with the final product demonstrating an HPLC purity exceeding 97% after simple workup procedures.
Q: Does this process require complex purification techniques like column chromatography?
A: No, the process is designed for industrial applicability, relying on standard extraction, washing, and concentration techniques rather than column chromatography, which facilitates easier separation and cost reduction in manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-methylbenzenesulfonic acid [(3-fluoro-oxetan-3-yl) methyl] ester Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the success of pharmaceutical development programs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements whether you are in early-stage clinical trials or full-scale commercial manufacturing. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify identity and potency. Our adoption of advanced synthetic methodologies, such as the one described in patent CN113461643A, demonstrates our dedication to continuous process improvement and our ability to provide cost-effective solutions for complex chemical challenges.
We invite you to engage with our technical procurement team to discuss how we can support your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how our optimized manufacturing processes can reduce your overall project costs. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your molecule, allowing you to make informed decisions about your supply chain strategy. Let us be your partner in bringing innovative therapies to market faster and more efficiently.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →