Scalable Continuous Production of Meropenem Intermediates for Global API Supply Chains
Introduction to Advanced Carbapenem Manufacturing
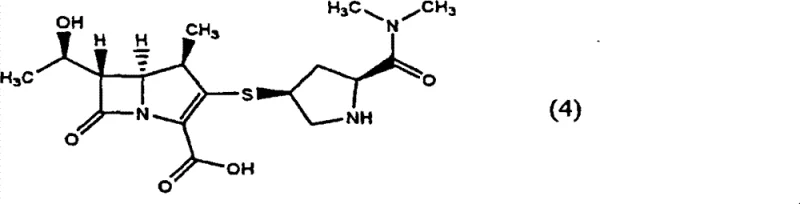
The pharmaceutical industry continuously seeks robust methodologies for producing beta-lactam antibiotics, particularly carbapenems, which are critical for treating severe bacterial infections. Patent CN101410398B introduces a significant technological advancement in the preparation of (4R, 5S, 6S)-3-[[(3S, 5S)-5-(dimethylaminocarbonyl)-3-pyrrolidinyl]thio]-6-[(1R)-1-hydroxyethyl]-4-methyl-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, widely known as Meropenem. This compound exhibits excellent antibacterial activity and represents a high-value target for generic API manufacturers. The core innovation lies in a streamlined process that eliminates the isolation and purification of the key synthetic intermediate, thereby addressing long-standing productivity issues in large-scale manufacturing. By integrating the coupling and deprotection steps into a continuous workflow, this method offers a compelling solution for enhancing throughput while maintaining stringent quality standards required for parenteral antibiotics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Meropenem has been plagued by inefficient downstream processing steps that hinder industrial scalability. Traditional routes typically involve the synthesis of a protected intermediate, often designated as compound (3), which requires rigorous isolation before proceeding to the final deprotection stage. In earlier methodologies, such as those described in Japanese Patent Laid-Open No. 60-104088, this isolation was achieved via column chromatography. While effective for laboratory-scale purification, column chromatography is notoriously difficult to implement in multi-ton commercial production due to high solvent consumption, low throughput, and operational complexity. Alternative approaches, referenced in international publication No. 2005/118586, attempted to replace chromatography with crystallization. However, these crystallization processes were excessively time-consuming, often requiring durations of 72 hours or more to achieve adequate separation. Furthermore, a critical chemical challenge arises from the nature of the reactants; the synthesis of intermediate (3) utilizes a thiol compound, residues of which are potent poisons for the palladium-on-carbon (Pd/C) catalysts used in the subsequent hydrogenation step. If the intermediate is not perfectly purified, catalyst activity drops precipitously, leading to incomplete reactions and increased impurity profiles.
The Novel Approach
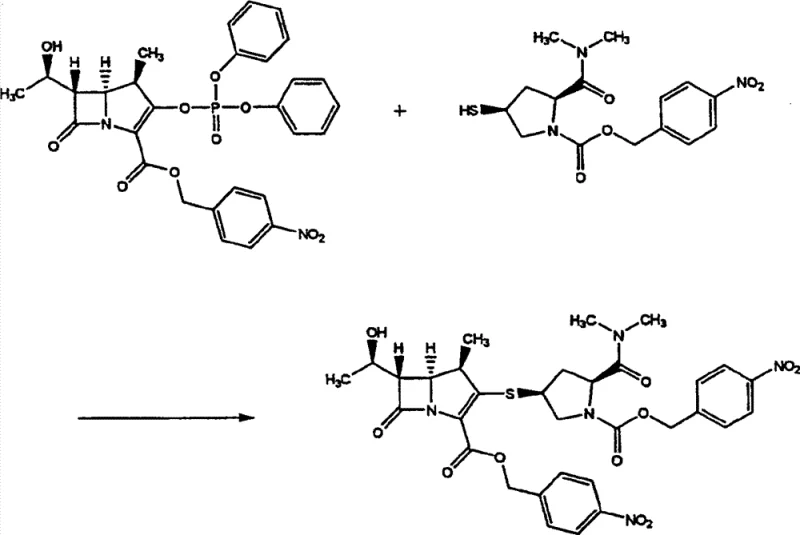
The methodology disclosed in CN101410398B fundamentally reengineers this workflow by adopting a telescoped strategy that bypasses the isolation of intermediate (3) entirely. Instead of separating the compound via chromatography or prolonged crystallization, the process utilizes the crude organic solvent solution of compound (3) directly in the hydrogenation reactor. This approach not only drastically reduces the cycle time by eliminating unit operations but also mitigates the risk of catalyst poisoning through careful solvent management and reaction condition optimization. The reaction involves coupling a phosphate ester derivative (Formula 1) with a mercapto-pyrrolidine derivative (Formula 2) in the presence of a base to generate the intermediate in situ. By maintaining the intermediate in solution, the process ensures a seamless transition to the deprotection phase, where protecting groups such as p-nitrobenzyl and p-nitrobenzyloxycarbonyl are removed simultaneously. This continuous operation significantly enhances the overall equipment effectiveness (OEE) of the manufacturing plant.
Mechanistic Insights into Telescoped Coupling and Hydrogenation
The chemical mechanism underpinning this process relies on a precise sequence of nucleophilic substitution followed by catalytic hydrogenolysis. In the first stage (Step A), the phosphate group at the C3 position of the carbapenem nucleus acts as a superior leaving group, facilitating nucleophilic attack by the thiol group of the pyrrolidine side chain. This reaction is typically conducted in polar aprotic solvents such as acetonitrile, N,N-dimethylformamide (DMF), or N-methylpyrrolidone (NMP), which stabilize the transition state and enhance reaction kinetics. The use of organic bases like N,N-diisopropylethylamine (DIPEA) or triethylamine is critical to scavenge the acidic byproducts generated during the substitution, ensuring the reaction proceeds to completion without degrading the sensitive beta-lactam ring. The resulting intermediate (3) retains two distinct protecting groups: a p-nitrobenzyl ester at the C2 carboxyl position and a p-nitrobenzyloxycarbonyl group on the pyrrolidine nitrogen. These groups are orthogonal to the beta-lactam stability but labile under hydrogenation conditions.
In the second stage (Step B), the deprotection mechanism involves the catalytic hydrogenation of the nitro-aromatic systems. When subjected to hydrogen gas in the presence of a palladium catalyst, the p-nitrobenzyl groups undergo hydrogenolysis to release the free carboxylic acid and amine functionalities, yielding the final Meropenem structure. A crucial aspect of this mechanism is the management of the reaction environment to prevent the decomposition of the carbapenem core, which is susceptible to hydrolysis. The patent highlights that maintaining the pH of the aqueous phase between 4 and 7 is essential; deviations outside this range accelerate degradation pathways. Additionally, the choice of solvent system plays a mechanistic role in solubilizing the intermediate while allowing the final product to partition effectively into the aqueous phase for isolation. The use of n-butanol in the hydrogenation mixture aids in this phase transfer and stabilizes the reaction mixture against thermal stress.
How to Synthesize Meropenem Efficiently
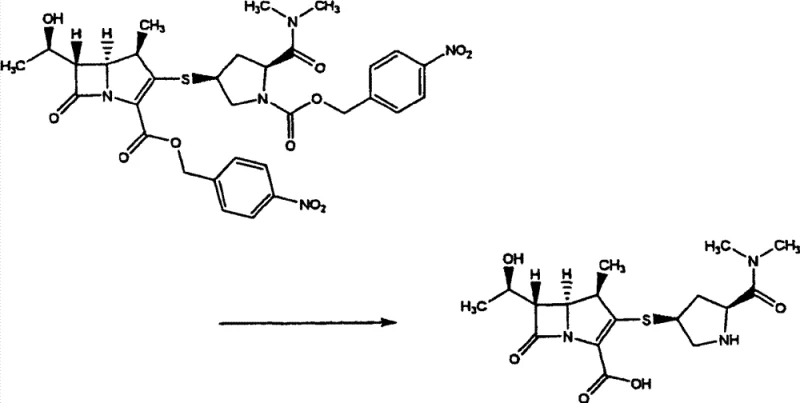
Implementing this continuous synthesis route requires careful attention to solvent selection and reaction parameters to ensure high yield and purity. The process begins with the dissolution of the phosphate starting material and the thiol coupling partner in a suitable organic solvent, followed by the controlled addition of base at low temperatures to manage exotherms. Once the coupling is complete, the reaction mixture can be subjected to a simple aqueous wash to remove inorganic salts before being transferred directly to the hydrogenation vessel. This operational simplicity is a key driver for adoption in GMP environments. For detailed standard operating procedures regarding stoichiometry, temperature ramps, and workup protocols, please refer to the technical guide below.
- React p-nitrobenzyl phosphate ester (Formula 1) with thiol pyrrolidine derivative (Formula 2) in an organic solvent like acetonitrile or DMF using a base such as DIPEA to form intermediate Formula 3.
- Without isolating Formula 3, subject the organic solvent solution directly to catalytic hydrogenation using Pd/C in a mixed solvent system containing water and an alcohol.
- Adjust the pH of the aqueous phase to between 4 and 7 during hydrogenation to prevent decomposition, then filter the catalyst and crystallize the final Meropenem hydrate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this telescoped synthesis route offers transformative benefits regarding cost structure and logistical reliability. By removing the isolation step for intermediate (3), manufacturers can significantly reduce the number of batch cycles required per month, effectively increasing capacity without capital expenditure on new reactors. The elimination of column chromatography removes a major bottleneck associated with solvent recovery and waste disposal, leading to substantial cost savings in raw material procurement and environmental compliance. Furthermore, the reduction in processing time from days to hours enhances the agility of the supply chain, allowing for faster response to market demand fluctuations for this critical antibiotic.
- Cost Reduction in Manufacturing: The primary economic driver of this technology is the removal of the intermediate isolation step. Conventional methods relying on crystallization require extended hold times exceeding 72 hours, tying up valuable reactor volume and utilities. By telescoping the reaction, the process eliminates these dormant periods, thereby maximizing asset utilization. Additionally, avoiding column chromatography removes the need for large volumes of silica gel and specialized eluents, which are significant cost centers in fine chemical manufacturing. The ability to use the crude reaction mixture directly in the next step also minimizes yield losses typically associated with mechanical transfers and filtration operations, leading to a higher overall mass balance and reduced cost of goods sold (COGS).
- Enhanced Supply Chain Reliability: Supply continuity for life-saving antibiotics like Meropenem is paramount. The conventional reliance on complex purification steps introduces multiple points of failure, such as filter clogging or crystallization inconsistencies, which can delay batch release. This novel process simplifies the workflow into two main chemical transformations, reducing the operational complexity and the likelihood of batch failures. The robustness of the hydrogenation step, when pH-controlled, ensures consistent quality output. This predictability allows supply chain planners to forecast production timelines with greater accuracy, reducing the need for excessive safety stock and minimizing the risk of shortages in the global pharmaceutical market.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process aligns with green chemistry principles by reducing solvent intensity. The removal of chromatographic purification significantly lowers the volume of organic waste generated per kilogram of product. Moreover, the process avoids the use of heavy metal scavengers often required to clean up residues from other catalytic methods, as the Pd/C catalyst can be easily filtered and potentially recycled. The scalability is inherent in the design; since the reaction does not rely on delicate crystallization kinetics for the intermediate, it can be scaled from pilot plants to multi-ton commercial reactors with minimal re-optimization, ensuring a smooth technology transfer for contract development and manufacturing organizations (CDMOs).
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation of this continuous carbapenem synthesis. These insights are derived directly from the experimental data and claims within the patent documentation, providing clarity on reaction conditions and quality control measures. Understanding these nuances is essential for R&D teams evaluating the feasibility of adopting this route for commercial production.
Q: Why is the isolation of intermediate compound (3) avoided in this process?
A: Isolating intermediate (3) traditionally requires column chromatography, which is difficult to scale industrially, or crystallization taking over 72 hours. Furthermore, residual thiol compounds from the synthesis of (3) can poison the palladium catalyst used in the subsequent deprotection step. By telescoping the steps, the process avoids these bottlenecks and catalyst poisoning issues.
Q: What solvents are optimal for the hydrogenation step in this carbapenem synthesis?
A: The patent specifies that the hydrogenation step (Step B) is preferably carried out in a mixed solvent system containing water and an organic solvent. Specifically, a solution containing an alcohol having 3 to 8 carbon atoms, such as n-butanol, is particularly preferred. A mixed solvent of n-butanol and ethyl acetate is cited as a highly effective system for this deprotection reaction.
Q: How is the stability of the carbapenem ring maintained during deprotection?
A: Stability is maintained by strictly controlling the pH of the aqueous phase during the hydrogenation reaction. The patent indicates that if the pH is higher than 7 or lower than 4, decomposition products tend to increase. Therefore, using a buffer solution to maintain the pH between 4 and 7 is critical for obtaining high-purity Meropenem.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Meropenem Supplier
As the global demand for broad-spectrum antibiotics continues to rise, securing a supply partner with deep technical expertise in carbapenem chemistry is essential. NINGBO INNO PHARMCHEM stands at the forefront of this sector, leveraging advanced continuous processing technologies to deliver high-quality intermediates and APIs. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the volumetric requirements of multinational pharmaceutical companies. We operate with stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch meets pharmacopeial standards.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific supply chain. By collaborating with us, you gain access to a Customized Cost-Saving Analysis that quantifies the economic impact of switching to this telescoped process. We encourage you to request specific COA data and route feasibility assessments to validate the compatibility of our manufacturing capabilities with your product portfolio. Let us drive efficiency and reliability in your Meropenem supply chain together.