Optimizing Armillarisin A Production: A Technical Breakthrough for Scalable Pharmaceutical Manufacturing
Optimizing Armillarisin A Production: A Technical Breakthrough for Scalable Pharmaceutical Manufacturing
The pharmaceutical industry continuously seeks robust, scalable, and cost-effective pathways for producing critical active pharmaceutical ingredients (APIs) and their intermediates. A significant advancement in this domain is detailed in patent CN114805273A, which outlines a novel preparation method for pharmaceutical-grade Armillarisin A, also known as Leupillin A. This coumarin derivative is pivotal in treating hepatobiliary disorders, including acute and chronic cholecystitis and viral hepatitis, due to its ability to promote bile secretion and regulate liver function. The patented process addresses long-standing challenges in the synthesis of this compound, specifically targeting the issues of low yield and poor purity that have plagued conventional manufacturing techniques. By leveraging a mild, base-catalyzed condensation reaction followed by an innovative purification protocol, this technology offers a compelling value proposition for reliable pharmaceutical intermediate supplier networks aiming to enhance their product portfolios with high-quality hepatoprotective agents.
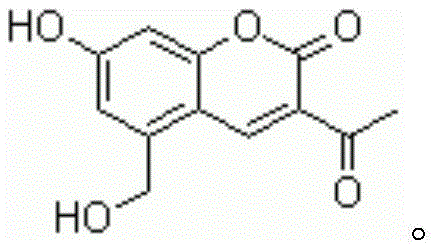
Armillarisin A, with the molecular formula C12H10O5 and a molecular weight of 234.2, presents specific physicochemical challenges, notably its insolubility in water and very slight solubility in ethanol, which historically complicated its isolation and purification. The breakthrough described in the patent data provides a streamlined solution that not only overcomes these solubility hurdles but also aligns with green chemistry principles by avoiding toxic catalysts and extreme reaction conditions. For R&D directors and procurement specialists alike, understanding the nuances of this synthesis route is critical for evaluating its potential impact on cost reduction in pharmaceutical intermediates manufacturing and ensuring a stable supply of this vital therapeutic agent.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of Armillarisin A has been hindered by inefficient extraction from natural sources or cumbersome synthetic routes that fail to deliver consistent quality at scale. Prior art, such as the methods referenced in existing literature, often suffers from yields hovering around 80%, which is suboptimal for commercial viability when dealing with high-value pharmaceutical compounds. Furthermore, traditional purification techniques struggle with the compound's inherent solubility profile; because Armillarisin A is practically insoluble in water and only sparingly soluble in common organic solvents like ethanol, recovering the product from reaction mixtures often leads to significant material loss. Conventional methods may also rely on harsh conditions or expensive catalysts that introduce heavy metal impurities, necessitating additional, costly downstream processing steps to meet stringent regulatory standards for pharmaceutical-grade materials. These inefficiencies translate directly into higher production costs and longer lead times, creating bottlenecks for commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology disclosed in CN114805273A introduces a refined synthetic pathway that maximizes efficiency through precise control of reaction parameters and a clever purification strategy. The process initiates with the condensation of 3,5-dihydroxybenzyl alcohol and ethyl ethoxymethylene acetoacetate in anhydrous ethanol, catalyzed by sodium ethoxide generated in situ from metallic sodium. This reaction proceeds under mild conditions without the need for high temperature or high pressure, significantly reducing energy consumption and equipment stress. The true innovation lies in the purification phase: the crude product is dissolved using ammonia water, which alters the solvent environment to a weakly alkaline state, drastically improving solubility and allowing for effective decolorization with activated carbon. Subsequent acidification with glacial acetic acid triggers precise crystallization, yielding a high-purity finished product. This approach not only simplifies the operational workflow but also ensures that the final API intermediate meets the rigorous specifications required by global regulatory bodies.
Mechanistic Insights into Base-Catalyzed Condensation and Purification
From a mechanistic perspective, the synthesis relies on a classic yet optimized condensation reaction facilitated by a strong base. The generation of sodium ethoxide serves as the primary catalytic driver, deprotonating the active methylene group of the ethyl ethoxymethylene acetoacetate to form a nucleophilic enolate. This species then attacks the electrophilic center of the 3,5-dihydroxybenzyl alcohol derivative, initiating the cyclization process that forms the core coumarin structure characteristic of Armillarisin A. The patent specifies a reaction time of 4 hours followed by a standing period of 12 hours, allowing the system to reach thermodynamic equilibrium and maximize the formation of the desired solid intermediate. The stoichiometry is carefully balanced, with a mass ratio of 3,5-dihydroxybenzyl alcohol to ethyl ethoxymethylene acetoacetate to sodium ethoxide optimized at approximately 1:1.5:4.7 to ensure complete conversion while minimizing side reactions. This precise control over molar ratios is crucial for maintaining a clean impurity profile, as excess reagents could lead to polymerization or the formation of difficult-to-remove byproducts.
The purification mechanism is equally sophisticated, addressing the specific solubility constraints of the target molecule. By introducing ammonia water, the process converts the phenolic hydroxyl groups of the coumarin structure into more soluble ammonium salts, effectively bringing the product into the solution phase where impurities can be adsorbed by activated carbon. This step is critical for removing colored impurities and trace organics that often persist in crude synthetic batches. The subsequent addition of glacial acetic acid reverses this solubility enhancement by lowering the pH to an acidic range (specifically adjusting to pH 2-3 in earlier steps and using acetic acid for final crystallization), causing the free phenolic form of Armillarisin A to precipitate out of the solution in a highly pure crystalline form. This pH-swing crystallization technique is a powerful tool for high-purity pharmaceutical intermediates production, as it leverages the intrinsic chemical properties of the molecule rather than relying on brute-force solvent evaporation or chromatography.
How to Synthesize Armillarisin A Efficiently
The synthesis of Armillarisin A via this patented route is designed for operational simplicity and reproducibility, making it an ideal candidate for technology transfer from the laboratory to the pilot plant and eventually to full-scale commercial production. The process begins with the preparation of Solution A by dissolving the starting materials in anhydrous ethanol, followed by the controlled addition of the sodium ethoxide catalyst. Critical process parameters include maintaining the reaction temperature within a moderate range and adhering to the specified stirring and standing times to ensure optimal crystal growth. The workup involves a series of filtration and decolorization steps that are standard in fine chemical manufacturing but are here applied with specific attention to pH control and solvent composition to maximize recovery. For a detailed breakdown of the standardized operating procedures, including specific mass ratios and temperature setpoints for each stage, please refer to the technical guide below.
- Dissolve 3,5-dihydroxybenzyl alcohol and ethyl ethoxymethylene acetoacetate in anhydrous ethanol, then react with freshly prepared sodium ethoxide.
- Filter the resulting solid, decolorize with activated carbon in purified water, and adjust pH to 2-3 to isolate the crude product.
- Purify the crude material by dissolving in ammonia-water/ethanol mixture, decolorizing again, and precipitating with glacial acetic acid at low temperature.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this novel synthesis route offers tangible strategic benefits that extend beyond mere technical feasibility. The elimination of high-temperature and high-pressure requirements fundamentally alters the risk profile of the manufacturing process, allowing for the use of simpler, more widely available reactor vessels rather than specialized high-pressure autoclaves. This reduction in equipment complexity translates directly into lower capital expenditure (CAPEX) and reduced maintenance overheads, facilitating a more agile response to market demand fluctuations. Furthermore, the reliance on commodity chemicals such as ethanol, metallic sodium, ammonia, and acetic acid ensures a robust and resilient supply chain, mitigating the risks associated with sourcing exotic or regulated reagents. The process's inherent safety profile, characterized by low toxicity and mild operating conditions, also reduces the regulatory burden and insurance costs associated with hazardous chemical manufacturing.
- Cost Reduction in Manufacturing: The economic implications of this process are profound, primarily driven by the significant improvement in yield and the simplification of the purification train. By achieving yields that approach theoretical maximums, the consumption of raw materials per kilogram of finished product is drastically minimized, directly lowering the variable cost of goods sold (COGS). Additionally, the avoidance of expensive transition metal catalysts eliminates the need for costly metal scavenging steps and the associated validation testing to ensure residual metals are within ppm limits. The energy efficiency of the process, operating without the need for intense heating or cooling cycles beyond standard crystallization temperatures, further contributes to substantial cost savings in utility consumption. These factors combine to create a highly competitive cost structure that allows suppliers to offer better pricing without compromising on margin.
- Enhanced Supply Chain Reliability: Supply chain continuity is paramount in the pharmaceutical sector, and this synthesis route enhances reliability through the use of stable, non-perishable raw materials. The reagents involved, such as 3,5-dihydroxybenzyl alcohol and ethyl ethoxymethylene acetoacetate, are commercially available from multiple global vendors, reducing the risk of single-source dependency. The robustness of the reaction conditions means that production schedules are less likely to be disrupted by equipment failures or safety incidents, ensuring consistent on-time delivery to downstream API manufacturers. Moreover, the simplified workflow reduces the overall cycle time from raw material intake to finished goods release, effectively reducing lead time for high-purity pharmaceutical intermediates and enabling a more responsive just-in-time inventory strategy.
- Scalability and Environmental Compliance: As environmental regulations become increasingly stringent, the green chemistry attributes of this process provide a significant compliance advantage. The method generates minimal waste, particularly by avoiding the heavy metal contamination typical of other catalytic systems, which simplifies wastewater treatment and disposal protocols. The use of ethanol as the primary solvent is favorable from an environmental, health, and safety (EHS) perspective, as it is less toxic and easier to recover and recycle compared to chlorinated or aromatic solvents. This alignment with sustainability goals not only future-proofs the manufacturing operation against tightening regulations but also appeals to environmentally conscious partners and stakeholders, reinforcing the supplier's reputation as a responsible manufacturer in the global fine chemicals market.
Frequently Asked Questions (FAQ)
To assist technical teams in evaluating the feasibility of integrating this technology into their existing operations, we have compiled answers to common inquiries regarding the synthesis and purification of Armillarisin A. These responses are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and relevance for process engineers and quality assurance professionals. Understanding these details is essential for conducting a thorough risk assessment and determining the resource allocation required for successful implementation.
Q: What are the key advantages of the new Armillarisin A preparation method over conventional processes?
A: The novel method described in CN114805273A eliminates the need for high-temperature and high-pressure reactions, utilizing mild conditions that significantly reduce equipment complexity and energy consumption while achieving superior yields compared to traditional extraction or synthesis routes.
Q: How does the purification process ensure high pharmaceutical grade quality?
A: The process employs a unique solubility-switch strategy using ammonia water to dissolve the crude product followed by precipitation with glacial acetic acid, which effectively removes impurities and ensures high purity without requiring complex chromatographic separation.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the reaction utilizes common, inexpensive reagents like ethanol and sodium metal, operates under ambient pressure, and generates minimal waste, making it highly scalable and compliant with modern environmental and safety standards for bulk manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Armillarisin A Supplier
The technological advancements detailed in patent CN114805273A represent a significant leap forward in the efficient production of hepatoprotective intermediates, yet realizing this potential requires a partner with deep technical expertise and proven manufacturing capabilities. NINGBO INNO PHARMCHEM stands ready to support your development goals, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with state-of-the-art reactors and rigorous QC labs capable of meeting stringent purity specifications, ensuring that every batch of Armillarisin A delivered meets the highest international standards for pharmaceutical applications. We understand the critical nature of supply chain stability and are committed to providing a seamless transition from process development to full-scale commercial supply.
We invite you to engage with our technical team to explore how this optimized synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this method. We encourage you to contact our technical procurement team today to索取 specific COA data and route feasibility assessments, allowing us to demonstrate our commitment to quality, efficiency, and partnership in the global pharmaceutical supply chain.