Scalable Synthesis Of Novel Antibacterial Urolithin B Derivatives For Pharmaceutical Applications
Scalable Synthesis Of Novel Antibacterial Urolithin B Derivatives For Pharmaceutical Applications
The escalating crisis of bacterial resistance, particularly the alarming statistic that 98% of Staphylococcus aureus strains are now resistant to penicillin, necessitates the urgent development of novel antimicrobial agents with distinct mechanisms of action. Patent CN109928963B introduces a groundbreaking synthetic methodology for producing three-carbon chain methyl piperidine urolithin B and its hydrochloride salt, addressing the critical limitation of poor water solubility inherent in the parent urolithin B molecule. This innovation leverages a strategic structural modification where a flexible 1,3-dibromopropane chain links the rigid urolithin B core to a polar methylpiperidine terminal, effectively transforming a difficult-to-handle natural product derivative into a viable pharmaceutical candidate. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediate suppliers, this patent outlines a robust, cost-effective pathway that bypasses complex enzymatic extraction in favor of efficient chemical synthesis.
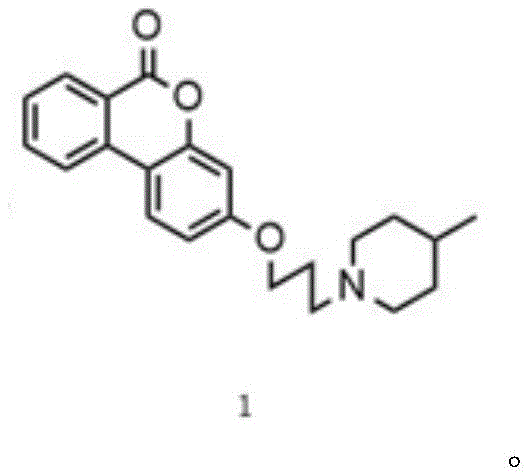
The structural integrity of the final compound, as depicted in the provided imagery, confirms the successful integration of the solubilizing tail while preserving the antibacterial pharmacophore. This molecular architecture is designed not only to enhance aqueous solubility for easier formulation but also to maintain potent activity against a broad spectrum of pathogens including Escherichia coli and Listeria monocytogenes. By shifting the production paradigm from extraction to total synthesis, manufacturers can achieve greater consistency in supply and significantly lower the cost of goods sold, making this a highly attractive target for cost reduction in antibacterial drug manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the utilization of urolithin B in therapeutic applications has been severely hampered by its physicochemical properties, specifically its extremely low water solubility which renders standard bioavailability assays and clinical formulations nearly impossible to execute effectively. Conventional attempts to modify the rigid urolithin scaffold often involve harsh conditions that risk degrading the sensitive lactone ring or require expensive, hard-to-source reagents that drive up production costs prohibitively. Furthermore, direct functionalization of the phenolic hydroxyl group without a spacer often results in steric hindrance issues, leading to low conversion rates and difficult purification profiles that generate excessive chemical waste. These historical bottlenecks have limited the commercial viability of urolithin-based drugs, forcing researchers to look for alternative scaffolds rather than optimizing this promising natural product.
The Novel Approach
The methodology disclosed in the patent overcomes these hurdles by employing a modular synthetic strategy that utilizes 1,3-dibromopropane as a flexible linker to bridge the gap between the hydrophobic core and the hydrophilic amine tail. This approach allows for mild reaction conditions, typically refluxing at 60°C in common solvents like acetone and acetonitrile, which minimizes energy consumption and equipment stress. The stepwise construction ensures that each transformation can be monitored and optimized independently, resulting in a cleaner reaction profile and higher overall yields compared to one-pot modifications. This streamlined process represents a significant advancement for the commercial scale-up of complex pharmaceutical intermediates, offering a clear route to high-purity active ingredients.
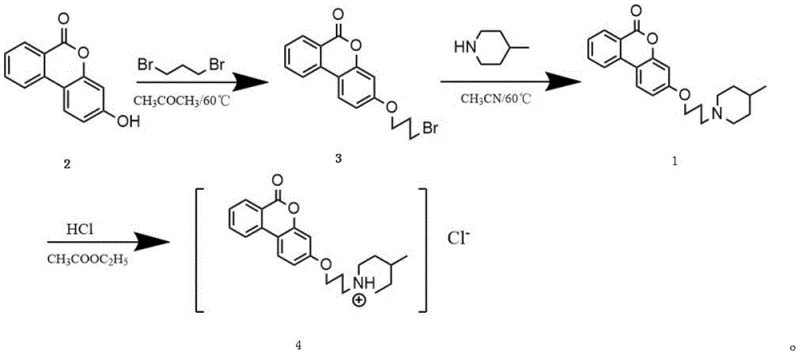
Mechanistic Insights into Nucleophilic Substitution and Salt Formation
The core of this synthesis relies on a classic yet highly effective nucleophilic substitution mechanism where the phenolic hydroxyl group of urolithin B acts as the nucleophile attacking the terminal carbon of 1,3-dibromopropane. In the presence of a base like potassium carbonate, the phenol is deprotonated to form a phenoxide ion, which then displaces a bromide ion to form the ether linkage, creating the bromopropylated intermediate. This intermediate retains a reactive bromine atom at the other end of the propyl chain, setting the stage for the subsequent amination step where 4-methylpiperidine attacks to displace the second bromide. This two-step alkylation-amination sequence is chemically elegant because it uses the dibromide as a bifunctional reagent, eliminating the need for activating agents or coupling reagents that would introduce additional impurities and cost.
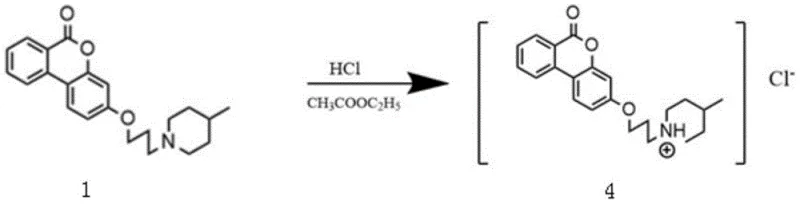
Following the formation of the free base, the process employs a straightforward acid-base salification reaction to generate the hydrochloride salt, which is crucial for enhancing the stability and crystallinity of the final drug substance. The mechanism involves dissolving the free base in ethyl acetate and introducing hydrogen chloride, which protonates the tertiary nitrogen of the piperidine ring to form a stable ammonium cation paired with a chloride anion. This ionic interaction drastically improves the lattice energy of the solid, facilitating precipitation and allowing for easy isolation via simple filtration. The ability to control particle size and morphology through the rate of acid addition provides an additional layer of process control essential for meeting stringent purity specifications required by regulatory bodies.
How to Synthesize Three-Carbon Chain Methyl Piperidine Urolithin B Efficiently
The synthesis protocol described in the patent offers a reproducible framework for laboratory and pilot-scale production, emphasizing precise stoichiometric control and thermal management to maximize yield. The process begins with the alkylation of urolithin B using a four-fold molar excess of 1,3-dibromopropane to drive the equilibrium forward, followed by a rigorous workup involving petroleum ether washing to remove unreacted alkylating agent. Subsequent amination with 4-methylpiperidine in acetonitrile requires careful TLC monitoring to ensure complete consumption of the bromo-intermediate before proceeding to purification. Detailed standardized synthesis steps see the guide below.
- Alkylate Urolithin B with 1,3-dibromopropane in acetone at 60°C to form the bromopropylated intermediate.
- React the bromopropylated intermediate with 4-methylpiperidine in acetonitrile at 60°C to introduce the soluble tail.
- Purify the free base via column chromatography and convert to the hydrochloride salt using HCl in ethyl acetate.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain perspective, this synthetic route offers substantial benefits by relying entirely on commodity chemicals that are readily available in the global market, thereby reducing the risk of raw material shortages. The elimination of transition metal catalysts or exotic reagents means that procurement teams can source inputs from multiple vendors, fostering competition and driving down input costs significantly. Furthermore, the use of standard solvents like acetone, acetonitrile, and ethyl acetate simplifies solvent recovery and recycling processes, contributing to a more sustainable and economically efficient manufacturing operation. These factors combined create a resilient supply chain capable of supporting long-term commercial production without the volatility associated with specialized reagents.
- Cost Reduction in Manufacturing: The process achieves cost optimization by utilizing inexpensive starting materials such as 1,3-dibromopropane and 4-methylpiperidine, which are produced on a massive industrial scale for other applications. By avoiding the need for chromatographic separation in the early stages and relying on simple filtration and crystallization for the final salt formation, the operational expenditure related to labor and consumables is drastically reduced. Additionally, the high atom economy of the substitution reactions ensures that a maximum proportion of the raw materials ends up in the final product, minimizing waste disposal costs.
- Enhanced Supply Chain Reliability: Because the synthesis does not depend on biologically derived extracts which can vary seasonally or geographically, the production timeline is predictable and consistent year-round. The robustness of the chemical steps allows for flexible batch sizing, meaning manufacturers can easily ramp up production volume to meet sudden spikes in demand without requalifying new suppliers or processes. This reliability is critical for maintaining continuous inventory levels of high-purity pharmaceutical intermediates, ensuring that downstream drug formulation is never interrupted by raw material delays.
- Scalability and Environmental Compliance: The reaction conditions are mild, operating at moderate temperatures around 60°C, which reduces the energy load on heating and cooling systems and lowers the carbon footprint of the facility. The waste streams generated are primarily organic solvents and inorganic salts, which are well-understood and easily treated using standard wastewater management protocols, ensuring compliance with increasingly strict environmental regulations. This ease of scale-up from gram to kilogram quantities without fundamental process changes makes it an ideal candidate for rapid technology transfer to commercial manufacturing sites.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this novel antibacterial agent, based on the detailed experimental data provided in the patent documentation. Understanding these nuances is vital for stakeholders evaluating the feasibility of integrating this compound into their existing drug development pipelines. The answers reflect the specific process parameters and quality control measures outlined in the intellectual property.
Q: What is the primary advantage of modifying Urolithin B with a methylpiperidine tail?
A: The primary advantage is significantly improved water solubility. Native Urolithin B has poor solubility which hinders biological testing and formulation. Adding the polar methylpiperidine group via a flexible carbon chain enhances bioavailability without destroying the rigid antibacterial parent ring.
Q: How is the purity of the intermediate controlled during synthesis?
A: Purity is strictly monitored using Thin Layer Chromatography (TLC). The reaction is stopped only when the fluorescent spots of the starting material disappear and the ratio of polar to non-polar spots stabilizes, ensuring minimal impurity carryover before column purification.
Q: Is this synthesis suitable for large-scale industrial production?
A: Yes, the process utilizes common solvents like acetone and acetonitrile and standard unit operations such as reflux, filtration, and column chromatography. The absence of sensitive catalysts or extreme conditions makes it highly amenable to commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Three-Carbon Chain Methyl Piperidine Urolithin B Supplier
As the demand for next-generation antibiotics grows, partnering with an experienced CDMO like NINGBO INNO PHARMCHEM ensures that your project benefits from our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, ensuring that every batch of three-carbon chain methyl piperidine urolithin B meets the highest international standards for safety and efficacy. We understand the critical nature of antibiotic development and are committed to providing a seamless transition from process development to full-scale manufacturing.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. By engaging with us early in your development cycle, you can secure specific COA data and route feasibility assessments that will accelerate your timeline to market. Let us leverage our expertise in fine chemical synthesis to help you bring this promising antibacterial candidate from the laboratory to the patients who need it most.