Advanced Palladium-Catalyzed Gamma-C(sp3)-H Thioether Synthesis for Scalable Pharmaceutical Manufacturing
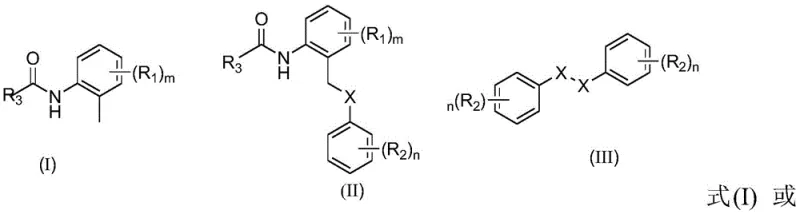
The pharmaceutical and fine chemical industries are constantly seeking more efficient and sustainable pathways to construct complex molecular architectures, particularly those containing sulfur heteroatoms which are ubiquitous in bioactive compounds. Patent CN111116461A introduces a groundbreaking methodology for the synthesis of palladium-catalyzed o-toluidine amide gamma-C-(sp3)H sulfur and selenide compounds. This technology represents a significant leap forward in C-H functionalization, specifically targeting the challenging activation of stable sp3 hybridized carbon-hydrogen bonds. By utilizing a pyridine-based directing group strategy, this process enables the direct coupling of o-toluidine derivatives with disulfides or diselenides under relatively mild thermal conditions ranging from 100 to 130°C. The innovation lies not only in the chemical transformation itself but in its ability to bypass the need for pre-functionalized substrates, thereby streamlining the synthetic route for high-value pharmaceutical intermediates and agrochemical building blocks.
Thioether motifs are critical structural elements found in numerous approved drugs, such as the atypical antipsychotic quetiapine, and serve as essential precursors for sulfone-based therapeutics. The ability to install these groups directly onto a carbon skeleton without prior halogenation offers substantial strategic advantages for process chemistry teams. This patent discloses a robust catalytic system employing palladium salts, organic peroxides as oxidants, and weak bases, which collectively facilitate the cleavage of the strong C-H bond and the subsequent formation of the C-S or C-Se bond. The broad substrate scope demonstrated in the experimental data suggests that this methodology is highly adaptable, accommodating various electronic and steric environments on both the amide and the disulfide components, making it a versatile tool for the reliable pharmaceutical intermediate supplier looking to diversify their catalog.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of thioether linkages has predominantly relied on nucleophilic substitution reactions between thiophenols and benzyl halides, such as benzyl bromide. While chemically straightforward, this classical approach suffers from severe drawbacks that hinder its applicability in modern green chemistry and large-scale manufacturing. The use of benzyl bromide introduces significant safety hazards due to its lachrymatory nature and potential carcinogenicity, requiring stringent containment measures and specialized waste disposal protocols. Furthermore, thiophenols are notoriously malodorous and toxic, posing occupational health risks to laboratory personnel and plant operators. From an efficiency standpoint, these traditional methods often generate stoichiometric amounts of salt waste and require the synthesis of halogenated precursors, which adds extra steps, reduces overall atom economy, and increases the cumulative cost of goods sold for the final active pharmaceutical ingredient.
The Novel Approach
In stark contrast, the methodology described in CN111116461A utilizes a direct C-H activation strategy that fundamentally alters the retrosynthetic logic for accessing these valuable scaffolds. By employing readily available o-toluidine amides and symmetrical disulfides, the process eliminates the need for hazardous alkylating agents and foul-smelling thiols. The reaction proceeds via a palladium-catalyzed cycle where the pyridine moiety acts as a transient directing group, guiding the metal center to the specific gamma-methyl C-H bond. This results in a highly selective transformation that minimizes side reactions and simplifies downstream purification. The operational simplicity is further enhanced by the use of common organic solvents like toluene and the ability to run the reaction under an air atmosphere in many instances, removing the need for expensive inert gas setups. This shift towards direct functionalization represents a paradigm shift in cost reduction in pharmaceutical intermediate manufacturing by reducing raw material complexity and waste treatment burdens.
Mechanistic Insights into Pd-Catalyzed Gamma-C(sp3)-H Activation
The core of this technological advancement lies in the intricate interplay between the palladium catalyst, the oxidant, and the directing group. The mechanism likely initiates with the coordination of the palladium species to the nitrogen atom of the pyridine ring in the o-toluidine amide substrate. This coordination brings the metal center into close proximity with the gamma-methyl group, facilitating the energetically demanding cleavage of the C(sp3)-H bond through a concerted metalation-deprotonation (CMD) pathway or a similar oxidative addition mechanism assisted by the acetate base. The resulting organopalladium intermediate is then intercepted by the disulfide reagent, which undergoes oxidative addition or ligand exchange to transfer the sulfur moiety to the carbon framework. The presence of tert-butyl hydroperoxide (TBHP) serves a dual role: it acts as a terminal oxidant to regenerate the active palladium species and may also assist in the homolytic cleavage of the disulfide bond to generate reactive sulfur radicals that participate in the catalytic cycle. This delicate balance of redox potentials ensures high turnover numbers and prevents catalyst deactivation.
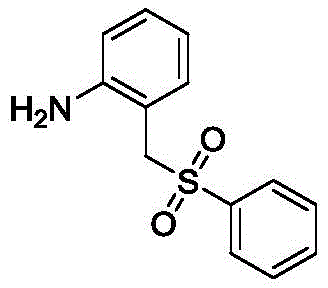
Furthermore, the versatility of this system extends beyond simple thioether formation, offering a gateway to higher oxidation state sulfur functionalities. The patent explicitly demonstrates that the resulting thioether products can be smoothly converted into sulfones through subsequent oxidation steps using reagents like m-chloroperoxybenzoic acid (m-CPBA). This downstream flexibility is crucial for medicinal chemists who often require sulfone analogs to modulate the metabolic stability and solubility profiles of drug candidates. The ability to access these oxidized derivatives from a common thioether intermediate simplifies the supply chain, allowing manufacturers to produce a library of related compounds from a single advanced intermediate. This modularity enhances the commercial viability of the process, as it allows for the production of high-purity OLED material precursors or pharmaceutical building blocks with minimal process changes, thereby maximizing asset utilization in multipurpose chemical plants.
How to Synthesize N-(o-tolyl)picolinamide Derivatives Efficiently
The practical implementation of this synthesis involves a straightforward protocol that is amenable to both laboratory discovery and pilot-scale production. The general procedure entails charging a reaction vessel with the o-toluidine amide substrate and the chosen disulfide coupling partner in a suitable aromatic solvent. Following the addition of the palladium catalyst, base, and oxidant, the mixture is heated to temperatures between 100 and 130°C for a duration of 8 to 24 hours. Reaction progress is typically monitored via thin-layer chromatography (TLC) to ensure complete conversion of the starting material. Upon completion, the workup involves cooling the reaction mixture, filtering off any insoluble solids, and concentrating the filtrate under reduced pressure. The crude product is then purified using standard silica gel column chromatography with a petroleum ether and ethyl acetate gradient to isolate the target gamma-C(sp3)-H thioether compound in high purity. For detailed standardized synthesis steps, please refer to the guide below.
- Dissolve N-(o-tolyl)picolinamide and diphenyl disulfide in toluene solvent within a reaction flask.
- Add palladium acetate catalyst, potassium acetate base, and tert-butyl hydroperoxide oxidant to the mixture.
- Stir the reaction at 120°C for 16 hours under air, then cool, filter, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this palladium-catalyzed C-H activation technology offers compelling economic and logistical benefits that directly impact the bottom line. By shifting away from pre-functionalized halogenated starting materials, companies can significantly reduce raw material procurement costs, as simple methyl-substituted anilines are generally cheaper and more abundant than their brominated or chlorinated counterparts. Additionally, the elimination of hazardous reagents like benzyl bromide reduces the regulatory burden associated with the storage and transport of controlled substances, leading to lower compliance costs and simplified logistics. The robustness of the reaction conditions, which tolerate air and moisture to a certain extent, further lowers the barrier to entry for contract manufacturing organizations (CMOs) that may not have specialized anaerobic capabilities, thereby expanding the pool of qualified suppliers and enhancing supply chain resilience against disruptions.
- Cost Reduction in Manufacturing: The economic advantage of this process is driven primarily by the simplification of the synthetic sequence. Traditional routes often require multi-step preparations to install leaving groups before sulfur incorporation, whereas this method achieves the transformation in a single catalytic step. This reduction in step count translates directly to lower labor costs, reduced solvent consumption, and decreased energy usage per kilogram of product. Furthermore, the high atom economy of using disulfides means that less mass is wasted as byproducts, minimizing the costs associated with waste disposal and environmental remediation. Although specific percentage savings depend on the specific substrate, the qualitative reduction in process complexity invariably leads to a more competitive cost structure for high-volume commercial production.
- Enhanced Supply Chain Reliability: Supply chain continuity is often threatened by the scarcity of specialized reagents or the volatility of prices for halogenated intermediates. This novel methodology relies on commodity chemicals such as toluene, potassium acetate, and commercially available disulfides, which are produced on a massive global scale. By decoupling the synthesis from niche, high-risk reagents, manufacturers can secure a more stable supply of critical inputs. The broad substrate scope also means that if one specific disulfide becomes unavailable, structurally similar alternatives can often be substituted with minimal process re-optimization. This flexibility ensures that production schedules remain intact even when facing market fluctuations, providing a reliable pharmaceutical intermediate supplier with a distinct competitive edge in fulfilling long-term contracts.
- Scalability and Environmental Compliance: Scaling C-H activation reactions has historically been challenging due to safety concerns regarding exotherms and oxidant handling. However, the conditions described in this patent utilize moderate temperatures and manageable oxidant loadings that are well-suited for batch reactor operations. The use of toluene, a solvent with well-established recovery and recycling protocols in the fine chemical industry, facilitates solvent management and reduces the environmental footprint of the process. Moreover, the avoidance of heavy metal waste streams associated with stoichiometric sulfur reagents aligns with increasingly stringent environmental regulations. The process generates fewer hazardous byproducts, simplifying the effluent treatment process and ensuring that the manufacturing facility remains compliant with local and international environmental standards, which is critical for maintaining operating licenses in regulated markets.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this palladium-catalyzed thioether synthesis technology. These answers are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity on reaction scope, safety profiles, and downstream applications. Understanding these nuances is essential for R&D teams evaluating this route for new drug discovery programs or process optimization initiatives.
Q: What are the safety advantages of this Pd-catalyzed thioether synthesis compared to traditional methods?
A: Traditional methods often rely on hazardous reagents like benzyl bromide and thiophenol, which pose significant toxicity and handling risks. This novel palladium-catalyzed approach utilizes stable disulfides and avoids pre-functionalized halogenated substrates, drastically improving operational safety and reducing hazardous waste generation in pharmaceutical intermediate manufacturing.
Q: Can this synthetic route be adapted for producing sulfone derivatives?
A: Yes, the resulting thioether products serve as versatile precursors. As demonstrated in the patent data, the thioether linkage can be efficiently oxidized using m-chloroperoxybenzoic acid to yield high-value sulfone compounds, which are critical scaffolds for bioactive molecules like 5-HT6 ligands used in treating attention deficit disorders.
Q: What represents the key technical breakthrough in this C-H functionalization strategy?
A: The primary breakthrough is the successful activation of the inert gamma-C(sp3)-H bond on the methyl group of o-toluidine derivatives. By employing a pyridine directing group and a palladium catalyst system, the method achieves high regioselectivity and atom economy without requiring harsh conditions or complex substrate preparation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-(o-tolyl)picolinamide Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H functionalization technologies in accelerating drug development timelines. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory discoveries can be seamlessly translated into industrial reality. Our state-of-the-art facilities are equipped to handle palladium-catalyzed reactions with the highest safety standards, and our rigorous QC labs enforce stringent purity specifications to meet the exacting demands of the global pharmaceutical market. We are committed to delivering high-purity thioether intermediates that serve as the foundation for next-generation therapeutics, leveraging our deep technical expertise to optimize yields and minimize impurities.
We invite potential partners to engage with our technical procurement team to discuss how this specific synthetic route can be tailored to your project needs. Whether you require a Customized Cost-Saving Analysis for an existing legacy process or need specific COA data for a novel analog, our experts are ready to provide comprehensive route feasibility assessments. By collaborating with us, you gain access to a supply chain that prioritizes quality, reliability, and continuous improvement, ensuring that your critical projects proceed without interruption. Contact us today to explore how our manufacturing capabilities can support your journey from benchtop to marketplace.