Advanced Synthetic Route for 1-Hydroxy-2-Trifluoromethyl-4-Iodo Pyridine Intermediates
Advanced Synthetic Route for 1-Hydroxy-2-Trifluoromethyl-4-Iodo Pyridine Intermediates
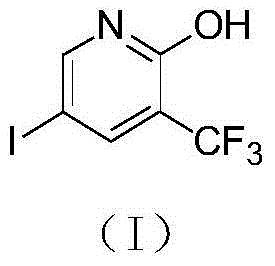
The pharmaceutical and agrochemical industries are constantly seeking robust, scalable, and environmentally compliant pathways for synthesizing complex heterocyclic building blocks. A pivotal development in this domain is detailed in patent CN103601671A, which discloses a highly efficient preparation method for 1-hydroxy-2-trifluoromethyl-4-iodo pyridine, a critical intermediate identified by CAS 887707-23-5. This compound serves as a versatile scaffold for constructing bioactive molecules, particularly in the realm of kinase inhibitors and metabolic modulators where the trifluoromethyl group enhances lipophilicity and metabolic stability. The disclosed methodology represents a significant leap forward in process chemistry, moving away from hazardous direct fluorination techniques toward a safer, step-wise functionalization of a pre-fluorinated pyridine ring. By leveraging a strategic sequence of oxidation, halogenation, hydrolysis, and microwave-assisted iodination, this process achieves exceptional purity and yield metrics that are essential for GMP-grade manufacturing. As a reliable pharmaceutical intermediate supplier, understanding the nuances of such patented routes allows us to offer clients not just a commodity, but a validated, risk-mitigated supply chain solution.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of fluorinated pyridine derivatives has been plagued by significant technical and economic hurdles that hinder large-scale adoption. Conventional routes often rely on direct electrophilic fluorination or the use of aggressive fluorinating agents like elemental fluorine or hydrogen fluoride, which pose severe safety risks regarding corrosion, toxicity, and explosion hazards. Furthermore, introducing an iodine atom at the specific 4-position of a pyridine ring without affecting other sensitive functional groups typically requires harsh conditions that can lead to extensive byproduct formation and difficult purification scenarios. These legacy methods frequently suffer from low atom economy, generating substantial amounts of hazardous waste that complicate environmental compliance and drive up disposal costs. Additionally, the reliance on cryogenic conditions or exotic catalysts in older protocols often results in inconsistent batch-to-batch reproducibility, creating bottlenecks for procurement managers who require steady, predictable supply volumes. The cumulative effect of these inefficiencies is a high cost of goods sold (COGS) and extended lead times, making traditional sources of such intermediates less attractive for cost-sensitive drug development programs.
The Novel Approach
In stark contrast, the novel approach outlined in the patent data utilizes 2-trifluoromethylpyridine as a stable, commercially available starting material, effectively bypassing the need for dangerous on-site fluorination steps. This strategy fundamentally shifts the risk profile of the synthesis, anchoring the process on well-understood heterocyclic transformations that are easier to control and scale. The route employs a logical progression where the pyridine nitrogen is first activated via oxidation, followed by a regioselective chlorination that sets the stage for subsequent nucleophilic substitution. This modular design allows for rigorous quality control at each intermediate stage, ensuring that impurities are removed before they can propagate through the synthesis. The final iodination step is particularly innovative, utilizing microwave irradiation to accelerate the reaction kinetics while maintaining mild thermal conditions, thereby preserving the integrity of the sensitive hydroxyl and trifluoromethyl groups. This holistic optimization results in a streamlined workflow that significantly reduces solvent consumption and energy usage, aligning perfectly with the goals of cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into the Four-Step Functionalization Strategy
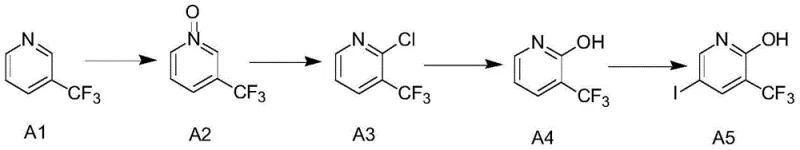
The core of this synthesis lies in a meticulously orchestrated four-step sequence that maximizes yield while minimizing side reactions. The process initiates with the oxidation of 2-trifluoromethylpyridine (Compound A1) using hydrogen peroxide in an organic acid medium, such as acetic acid, at temperatures ranging from 60°C to 100°C. This step generates the corresponding N-oxide (Compound A2), which activates the pyridine ring towards nucleophilic attack at the 2-position. Following isolation, the N-oxide undergoes chlorination with phosphorus oxychloride (POCl3) at elevated temperatures between 80°C and 120°C. This transformation replaces the oxygen of the N-oxide with a chlorine atom, yielding 2-chloro-3-trifluoromethylpyridine (Compound A3), a key electrophilic precursor. The third stage involves a hydrolysis reaction where the chloro group is displaced by a hydroxyl group using a strong base like sodium hydroxide or potassium hydroxide in an alcoholic solvent at 70°C to 120°C. This nucleophilic aromatic substitution is highly efficient, converting the chloro-intermediate into 1-hydroxy-2-trifluoromethylpyridine (Compound A4) with excellent conversion rates. Finally, the introduction of the iodine moiety is achieved using N-iodosuccinimide (NIS) under microwave heating conditions at 70°C to 100°C. This final electrophilic substitution selectively targets the 4-position of the pyridine ring, driven by the electronic activation provided by the adjacent hydroxyl and trifluoromethyl groups, resulting in the target molecule (Compound A5) with high regioselectivity.
Impurity control is embedded deeply within the mechanistic design of this pathway, ensuring the delivery of high-purity pharmaceutical intermediates. Each reaction step is followed by a robust workup procedure involving pH adjustment, solvent extraction, and recrystallization, which systematically removes unreacted starting materials, inorganic salts, and organic byproducts. For instance, the use of sodium sulfite solution to neutralize excess oxidant in the first step prevents oxidative degradation of the product in subsequent stages. Similarly, the careful control of pH during the hydrolysis and extraction phases ensures that the phenolic product remains in the organic phase while inorganic impurities are washed away into the aqueous layer. The final recrystallization from alcohol solvents serves as a polishing step, eliminating trace isomers and residual succinimide from the iodination step. This multi-layered purification strategy guarantees that the final API intermediate meets stringent specifications for heavy metals, residual solvents, and related substances, which is critical for downstream coupling reactions in drug synthesis.
How to Synthesize 1-Hydroxy-2-Trifluoromethyl-4-Iodo Pyridine Efficiently
Executing this synthesis requires precise adherence to the reaction parameters defined in the patent to ensure optimal yield and safety. The process is designed to be scalable, moving seamlessly from laboratory benchtop quantities to multi-ton production campaigns without losing efficiency. Operators must carefully monitor temperature ramps, especially during the exothermic addition of hydrogen peroxide and phosphorus oxychloride, to prevent thermal runaways. The use of microwave reactors for the final iodination step is a key differentiator, offering rapid heating and uniform energy distribution that conventional oil baths cannot match. Detailed standard operating procedures (SOPs) should include specific guidelines for solvent recovery and recycling, particularly for chlorinated solvents like dichloromethane and chlorobenzene used in the extraction phases. Below is the structured guide for the standardized synthesis steps.
- Oxidize 2-trifluoromethylpyridine with hydrogen peroxide in organic acid at 60-100°C to form the N-oxide intermediate.
- React the N-oxide with phosphorus oxychloride at 80-120°C to introduce the chlorine atom at the 2-position.
- Perform alkaline hydrolysis using sodium or potassium hydroxide at 70-120°C to convert the chloro-group to a hydroxyl group.
- Conduct the final iodination using N-iodosuccinimide under microwave heating at 70-100°C to yield the target pyridine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route translates into tangible strategic benefits that extend beyond simple unit pricing. The primary advantage lies in the drastic simplification of the supply chain; by utilizing 2-trifluoromethylpyridine as a feedstock, manufacturers avoid the volatility and regulatory burdens associated with sourcing elemental fluorine or managing complex fluorination reactors. This stability in raw material sourcing ensures consistent production schedules and mitigates the risk of supply disruptions caused by upstream chemical shortages. Furthermore, the high yield at every stage—reported to be substantially above 90% for individual steps—means that less raw material is wasted, directly lowering the effective cost per kilogram of the final product. This efficiency allows suppliers to offer more competitive pricing structures without compromising on quality margins, providing a clear path for cost reduction in API manufacturing for our partners.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts and the avoidance of cryogenic conditions significantly lower the operational expenditure (OPEX) associated with this synthesis. Traditional methods often require palladium or copper catalysts for halogenation, which not only add to the material cost but also necessitate expensive metal scavenging steps to meet regulatory limits for residual metals in pharmaceuticals. By relying on reagents like hydrogen peroxide, phosphorus oxychloride, and N-iodosuccinimide, this route utilizes cost-effective, commodity chemicals that are readily available in the global market. Additionally, the high atom economy of the microwave-assisted iodination step reduces energy consumption, further contributing to a leaner, more cost-efficient production model that maximizes return on investment for large-scale batches.
- Enhanced Supply Chain Reliability: The robustness of this synthetic pathway ensures a reliable supply of critical intermediates, which is paramount for maintaining continuous drug manufacturing operations. The process tolerances are wide enough to accommodate minor variations in raw material quality without impacting the final product specification, reducing the likelihood of batch failures and production delays. Since the starting material, 2-trifluoromethylpyridine, is a stable liquid that can be stored and transported safely, inventory management becomes simpler and less risky compared to handling gaseous or highly reactive fluorinating agents. This reliability allows procurement teams to negotiate longer-term contracts with confidence, knowing that the supplier has a proven, resilient method for commercial scale-up of complex pharmaceutical intermediates that can withstand market fluctuations.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this route is designed for sustainability, addressing the growing pressure on chemical companies to reduce their carbon footprint. The process generates minimal hazardous waste, as the byproducts are primarily inorganic salts and water-soluble organics that can be treated using standard wastewater facilities. The absence of heavy metal catalysts simplifies the disposal of mother liquors and reduces the environmental liability associated with toxic sludge. Moreover, the scalability of the microwave-assisted step has been demonstrated to be feasible for industrial applications, allowing for reducing lead time for high-purity intermediates by accelerating the slowest step in the synthesis. This combination of safety, scalability, and environmental stewardship makes the process highly attractive for multinational corporations with strict supplier codes of conduct.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this specialized pyridine derivative. These insights are derived directly from the technical specifications and advantageous effects described in the patent literature, providing clarity for R&D and procurement stakeholders. Understanding these details helps in assessing the feasibility of integrating this intermediate into your specific drug discovery or process development pipelines.
Q: Why is 2-trifluoromethylpyridine chosen as the starting material instead of direct fluorination?
A: Using 2-trifluoromethylpyridine as the parent scaffold avoids the hazardous and complex direct fluorination processes often required in traditional synthesis. This strategy significantly enhances process safety and reduces the generation of toxic fluoride waste, aligning with modern green chemistry principles while maintaining high structural integrity of the trifluoromethyl group.
Q: What are the advantages of using microwave heating in the final iodination step?
A: The implementation of microwave heating in the final substitution step drastically reduces reaction time and energy consumption compared to conventional thermal heating. This technology allows for precise temperature control between 70°C and 100°C, minimizing side reactions and decomposition, which ultimately leads to superior purity profiles and higher isolated yields of the iodo-substituted product.
Q: How does this synthesis route impact the overall cost of pharmaceutical intermediate manufacturing?
A: This route offers substantial cost reductions by utilizing readily available commercial starting materials and avoiding expensive transition metal catalysts. The high yield at each stage, consistently exceeding 90%, minimizes raw material waste and downstream purification costs, making it an economically viable solution for large-scale commercial production of complex heterocyclic intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Hydroxy-2-Trifluoromethyl-4-Iodo Pyridine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of new therapeutic agents depends heavily on the quality and consistency of the starting materials. Our technical team has thoroughly analyzed the synthetic route described in CN103601671A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. We are equipped with state-of-the-art reactor systems capable of handling the specific temperature and pressure requirements of this process, including specialized microwave reactors for the final iodination step. Our commitment to quality is underscored by our stringent purity specifications and rigorous QC labs, which utilize advanced analytical techniques to verify the identity and purity of every batch, ensuring it meets the exacting standards of the global pharmaceutical industry.
We invite you to collaborate with us to leverage this advanced synthesis technology for your upcoming projects. By partnering with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements, helping you optimize your budget without sacrificing quality. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Whether you are in the early stages of drug discovery or preparing for clinical trial material production, our expertise in fluorinated heterocycles ensures that you have a dependable partner dedicated to accelerating your timeline to market.