Advanced Synthesis of Dabigatran Etexilate Impurity Standards for Global QC Compliance
The pharmaceutical industry's relentless pursuit of safety and efficacy mandates rigorous quality control protocols, particularly for potent anticoagulants like Dabigatran etexilate. Patent CN102964307A introduces a groundbreaking methodology for the preparation of a critical related substance, specifically 2-{[4-(amino-n-hexyloxyimido-methylene)-phenylimino]-methylene}-1-methyl-1hydro-benzimidazole-5-ethyl formate. This compound serves as an essential reference standard for identifying and quantifying impurities during the manufacturing of the active pharmaceutical ingredient. The ability to synthesize this specific structural analog with high fidelity allows quality assurance teams to establish precise detection limits, thereby safeguarding patient health against potential toxicological risks associated with process by-products. 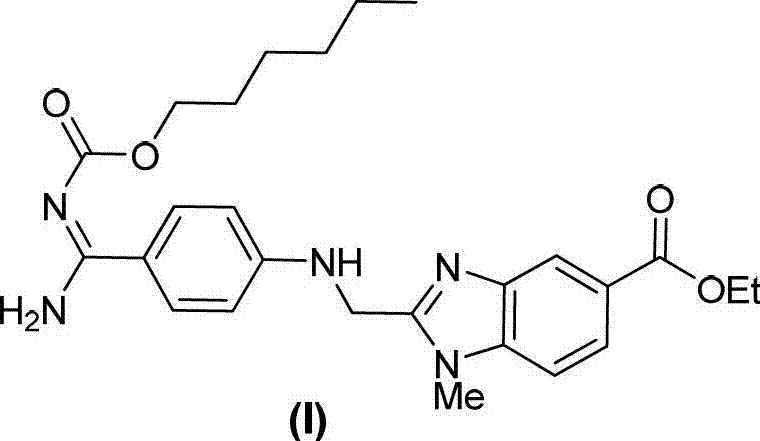 By securing a stable supply of such certified reference materials, pharmaceutical manufacturers can ensure their final products meet the stringent regulatory requirements set forth by global health authorities, ultimately reinforcing the integrity of the therapeutic supply chain.
By securing a stable supply of such certified reference materials, pharmaceutical manufacturers can ensure their final products meet the stringent regulatory requirements set forth by global health authorities, ultimately reinforcing the integrity of the therapeutic supply chain.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, obtaining specific impurity standards for complex molecules like Dabigatran etexilate has been a significant bottleneck for analytical laboratories and production facilities. In many conventional scenarios, these related substances are merely observed as trace by-products during the main API synthesis, making their isolation in sufficient quantities for analytical method validation extremely difficult and cost-prohibitive. Relying on isolation from crude reaction mixtures often results in low yields and inconsistent purity levels, which compromises the accuracy of high-performance liquid chromatography (HPLC) assays. Furthermore, without a dedicated synthetic route, there is no guarantee of the structural identity of the isolated impurity, leading to potential false positives or negatives in quality control testing. This uncertainty forces manufacturers to rely on expensive external sourcing or accept suboptimal detection capabilities, which poses a risk to batch release consistency and regulatory compliance.
The Novel Approach
The methodology disclosed in the patent data presents a robust and deliberate synthetic strategy designed to overcome the scarcity and inconsistency of traditional impurity sourcing. Instead of relying on chance isolation, this approach constructs the target molecule through a logical sequence of chemical transformations starting from readily available precursors. The process involves a controlled acid-catalyzed reaction followed by a selective ammonification and a final coupling step, ensuring that the desired structural features are introduced with high specificity. This targeted synthesis eliminates the variability associated with isolation techniques, providing a reproducible source of the related substance. By decoupling the production of the impurity standard from the main API manufacturing line, companies can maintain independent quality control benchmarks, ensuring that any deviation in the main process is detected with maximum sensitivity and reliability.
Mechanistic Insights into Acid-Catalyzed Esterification and Amidination
The core of this synthetic innovation lies in the precise manipulation of functional groups to construct the unique imino-methylene bridge characteristic of the target molecule. The initial step involves the reaction of a benzimidazole precursor under acidic conditions in the presence of an alcohol, which facilitates the formation of an ethoxy imino intermediate. This transformation is critical as it activates the molecular framework for subsequent nucleophilic attacks, setting the stage for the introduction of the nitrogen-containing side chain. The use of specific acids, such as hydrogen chloride, ensures the formation of a stable hydrochloride salt, which simplifies the isolation of the intermediate as a solid precipitate. This salt formation not only drives the reaction equilibrium forward but also protects sensitive functional groups from degradation during the workup phase, preserving the integrity of the molecular scaffold for the next transformation.
Following the formation of the ethoxy imino intermediate, the process proceeds through an ammonification reaction where the ethoxy group is displaced by an amino group. This step is pivotal for generating the amidine functionality required for the final coupling reaction. The use of ammonia or ammonium salts in a suitable solvent system allows for a clean substitution reaction, minimizing the formation of side products that could complicate downstream purification. The resulting amidine hydrochloride is then reacted with n-hexyl chloroformate in the presence of a tertiary amine base, such as triethylamine, to form the final carbamate linkage. 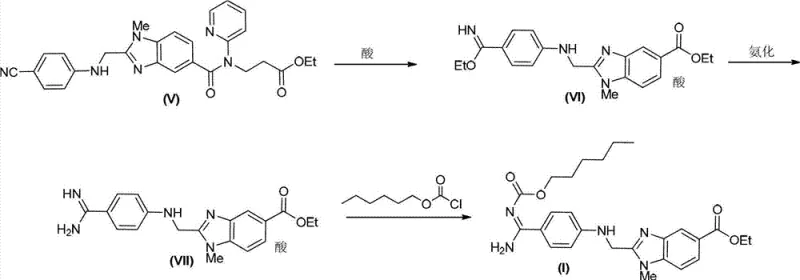 This final acylation step is carefully controlled to prevent over-reaction or hydrolysis, ensuring that the n-hexyloxyimido moiety is installed with high fidelity. The entire mechanistic pathway is designed to maximize atom economy and minimize waste, reflecting a modern approach to fine chemical synthesis that prioritizes both efficiency and environmental responsibility.
This final acylation step is carefully controlled to prevent over-reaction or hydrolysis, ensuring that the n-hexyloxyimido moiety is installed with high fidelity. The entire mechanistic pathway is designed to maximize atom economy and minimize waste, reflecting a modern approach to fine chemical synthesis that prioritizes both efficiency and environmental responsibility.
How to Synthesize Dabigatran Etexilate Related Substance Efficiently
Implementing this synthesis route requires careful attention to reaction conditions and stoichiometry to achieve the reported purity levels. The process begins with the dissolution of the starting material in ethanol, followed by the slow introduction of acid gas to initiate the esterification. Once the intermediate is isolated, it is subjected to ammonification in a solvent like ethanol or methanol, where temperature and pressure must be monitored to ensure complete conversion. The final step involves the addition of the chloroformate reagent under basic conditions, typically in a halogenated solvent, followed by a rigorous purification protocol.
- React the precursor benzimidazole derivative with acid and alcohol to form the ethoxy imino intermediate hydrochloride salt.
- Perform an ammonification reaction on the intermediate using ammonia gas or ammonium salts to generate the amidine hydrochloride.
- Couple the amidine intermediate with n-hexyl chloroformate in the presence of a base to yield the final target related substance.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers substantial strategic benefits beyond mere technical feasibility. The reliance on commodity chemicals and standard solvents significantly reduces the complexity of raw material sourcing, mitigating the risk of supply disruptions caused by specialized reagent shortages. Since the process does not require exotic catalysts or extreme reaction conditions, it can be implemented in existing multipurpose chemical manufacturing facilities without the need for capital-intensive equipment upgrades. This flexibility translates directly into lower operational expenditures and faster time-to-market for the reference standards, allowing pharmaceutical companies to respond more agilely to regulatory changes or production issues. Furthermore, the robustness of the synthesis ensures a consistent supply of high-quality material, which is essential for maintaining uninterrupted quality control operations across global manufacturing sites.
- Cost Reduction in Manufacturing: The elimination of complex isolation procedures and the use of inexpensive, widely available reagents drastically lower the cost of goods sold for these critical reference standards. By avoiding the need for preparative HPLC purification of trace impurities from API batches, manufacturers can achieve significant savings in solvent consumption and labor hours. The high yield of the final step further contributes to cost efficiency, ensuring that every kilogram of starting material is converted into valuable product with minimal waste. This economic advantage allows companies to allocate resources more effectively towards other critical areas of drug development and production.
- Enhanced Supply Chain Reliability: The modular nature of this three-step synthesis allows for decentralized production, reducing the dependency on single-source suppliers for finished reference standards. By licensing or implementing this technology internally, pharmaceutical firms can secure their own supply of impurity markers, insulating themselves from external market volatility. The stability of the intermediates also permits batch storage, enabling companies to build strategic reserves that buffer against unexpected demand spikes or logistical delays. This level of supply chain autonomy is crucial for maintaining continuous manufacturing operations and meeting strict regulatory submission deadlines.
- Scalability and Environmental Compliance: The process is inherently scalable, having been demonstrated to work efficiently from gram to kilogram scales without loss of yield or purity. The use of standard organic solvents facilitates efficient recovery and recycling systems, aligning with modern green chemistry principles and reducing the environmental footprint of the manufacturing process. Additionally, the absence of heavy metal catalysts simplifies waste treatment protocols and lowers the cost of environmental compliance. This sustainability profile not only meets corporate social responsibility goals but also future-proofs the manufacturing process against increasingly stringent environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. Understanding these details is crucial for R&D and procurement teams evaluating the feasibility of adopting this method for their internal quality control workflows.
Q: What is the primary advantage of this synthesis route for quality control?
A: This method provides a reliable pathway to synthesize specific related substances that are difficult to isolate from the main API production stream, ensuring accurate HPLC calibration and impurity profiling.
Q: Are the reagents used in this process commercially scalable?
A: Yes, the process utilizes common industrial solvents like ethanol and methylene dichloride, along with standard reagents such as triethylamine and chloroformic acid esters, facilitating easy scale-up.
Q: How does this method impact the purity of the final reference standard?
A: The inclusion of a column chromatography purification step in the final stage ensures that the resulting solid meets stringent purity specifications required for analytical validation.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dabigatran Etexilate Related Substance Supplier
At NINGBO INNO PHARMCHEM, we understand the critical role that high-purity reference standards play in ensuring the safety and efficacy of life-saving medications. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of material we deliver meets the highest international standards. Our commitment to quality extends beyond the product itself, encompassing comprehensive documentation and support to facilitate your regulatory filings and internal validations.
We invite you to collaborate with us to optimize your supply chain for Dabigatran etexilate related substances and other critical pharmaceutical intermediates. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific production volumes. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can enhance your operational efficiency and reduce overall costs.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →