Advanced Ripretinib Manufacturing: A Technical Breakthrough for Scalable API Production
Advanced Ripretinib Manufacturing: A Technical Breakthrough for Scalable API Production
The pharmaceutical landscape for Gastrointestinal Stromal Tumors (GIST) treatment has been significantly advanced by the introduction of Ripretinib, a potent switch-control kinase inhibitor. As the demand for this critical Active Pharmaceutical Ingredient (API) grows globally, the efficiency and safety of its manufacturing process become paramount for supply chain stability. Patent CN114213411B, published in mid-2023, introduces a transformative synthetic methodology that addresses the longstanding bottlenecks of traditional production routes. This technical insight report analyzes the novel five-step synthesis pathway, highlighting its superior atom economy, enhanced safety profile through the elimination of hazardous reagents, and robust scalability. For R&D directors and procurement strategists, understanding these mechanistic improvements is essential for securing a reliable API intermediate supplier capable of delivering high-purity materials consistently.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis routes, such as those disclosed in WO2013184119A1 and CN112625038A, have historically plagued manufacturers with significant operational and environmental challenges. These legacy processes typically rely on a convergent strategy that involves the separate preparation of complex intermediates followed by a difficult coupling step. A critical flaw in these conventional methods is the reliance on phosphorus oxychloride for chlorination and manganese dioxide for oxidation, reactions that generate substantial volumes of acidic and heavy metal-contaminated wastewater, posing severe environmental compliance burdens. Furthermore, the final assembly of the urea moiety often necessitates the use of phenyl isocyanate, a reagent known for its extreme toxicity, pungent odor, and stringent handling requirements, which complicates worker safety protocols and increases facility containment costs.
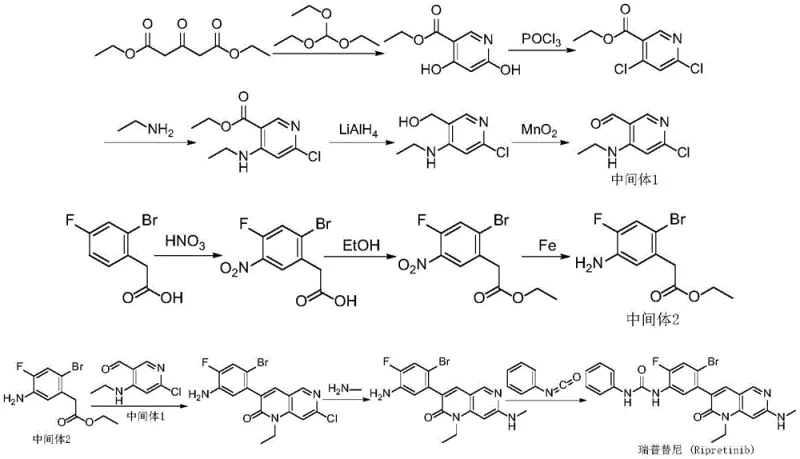
Beyond the environmental and safety hazards, the conventional pathways suffer from poor process efficiency due to their excessive length and harsh reaction conditions. The prior art routes often require extended reaction times, with some critical steps taking up to four days to reach completion, drastically reducing throughput capacity and increasing energy consumption. The multi-step nature of these older syntheses also inherently accumulates impurities, necessitating rigorous and costly purification procedures to meet the stringent quality standards required for oncology drugs. Consequently, these factors combine to create a high-cost manufacturing baseline that limits the ability of suppliers to offer competitive pricing while maintaining robust inventory levels for global distribution networks.
The Novel Approach
In stark contrast to the cumbersome legacy methods, the invention detailed in CN114213411B proposes a streamlined, linear synthesis strategy that fundamentally reimagines the construction of the naphthyridinone core. This novel approach initiates with a highly efficient substitution reaction between readily available starting materials, methyl 2-bromo-4-fluoro-5-nitrobenzoate and 4-amino-6-chloro-3-pyridinecarbaldehyde, bypassing the need for pre-functionalized complex intermediates. By integrating the ring-closing event directly into a catalytic hydrogenation step, the new process eliminates the need for stoichiometric oxidants like manganese dioxide and harsh chlorinating agents, thereby significantly reducing the generation of hazardous waste streams. This shift not only aligns with green chemistry principles but also simplifies the downstream workup procedures, allowing for faster cycle times and improved overall equipment effectiveness in a commercial plant setting.
Mechanistic Insights into Reductive Cyclization and Urea Formation
The cornerstone of this innovative synthesis lies in the elegant execution of the reductive cyclization step, which simultaneously reduces the nitro group and facilitates the intramolecular condensation to form the 1,6-naphthyridin-2(1H)-one scaffold. Utilizing a heterogeneous catalyst system, specifically 10% Palladium on Carbon (Pd/C), under mild hydrogen pressure allows for precise control over the reduction potential, preventing the over-reduction of other sensitive functional groups such as the aryl bromide. This chemoselectivity is crucial for maintaining the integrity of the molecular architecture required for subsequent functionalization. The reaction proceeds in ethanol, a benign and easily recyclable solvent, further enhancing the environmental profile of the process while ensuring high solubility of the intermediates to drive the equilibrium towards the desired cyclic product with exceptional conversion rates.
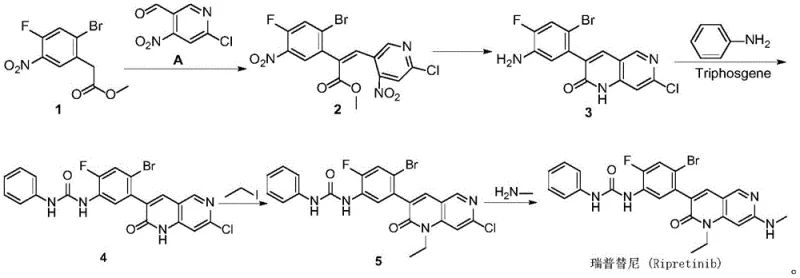
Another critical mechanistic advancement is the strategic modification of the urea bond formation, which traditionally poses the highest safety risk in Ripretinib synthesis. Instead of employing the hazardous phenyl isocyanate, this patent discloses an in-situ generation of the reactive isocyanate species using aniline and triphosgene in the presence of an organic base like triethylamine. This method effectively masks the toxicity of the isocyanate by generating it only in the immediate presence of the nucleophile, thereby minimizing exposure risks and eliminating the need for storing bulk quantities of dangerous reagents. The reaction occurs smoothly in dichloromethane at ambient temperatures, demonstrating high regioselectivity for the aniline nitrogen over other potential nucleophilic sites on the naphthyridinone ring, which ensures the formation of the correct regioisomer with minimal byproduct formation and simplifies the final purification burden.
How to Synthesize Ripretinib Efficiently
The implementation of this patented methodology requires careful attention to reaction parameters to maximize the reported yields and purity profiles. The process begins with the activation of the pyridine aldehyde derivative followed by a controlled hydrogenation that demands precise catalyst loading to ensure complete cyclization without dehalogenation. Following the core construction, the sequential alkylation and amination steps must be managed to prevent over-alkylation or hydrolysis of the urea linkage. Detailed standard operating procedures regarding temperature gradients, addition rates, and quenching protocols are essential for reproducing the laboratory success on a pilot or commercial scale. For a comprehensive breakdown of the specific molar ratios, solvent choices, and thermal profiles required for each transformation, please refer to the standardized synthesis guide below.
- Perform a base-catalyzed substitution reaction between methyl 2-bromo-4-fluoro-5-nitrobenzoate and 4-amino-6-chloro-3-pyridinecarbaldehyde in THF at 50°C.
- Execute a catalytic hydrogenation and reductive cyclization using 10% Pd/C in ethanol to form the naphthyridinone core structure.
- React the intermediate with aniline and triphosgene in dichloromethane to safely construct the urea linkage without using toxic phenyl isocyanate.
- Conduct an alkylation reaction with ethyl iodide and potassium carbonate in DMF, followed by a final amination with methylamine to yield Ripretinib.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route translates directly into tangible operational efficiencies and risk mitigation strategies. The primary economic driver is the drastic simplification of the synthetic sequence, which reduces the number of unit operations and the associated labor, utility, and equipment occupancy costs. By shortening the overall production timeline and eliminating the four-day reaction hold times characteristic of older methods, manufacturers can significantly increase their annual production capacity without requiring additional capital investment in new reactor trains. This enhanced throughput capability ensures a more reliable supply of high-purity API intermediates, mitigating the risk of stockouts that can disrupt downstream drug formulation and patient access.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous reagents such as phenyl isocyanate and manganese dioxide removes significant cost centers related to specialized storage, handling, and waste disposal. Furthermore, the high yields achieved in each step, particularly the near-quantitative conversion in the initial substitution and hydrogenation stages, minimize the loss of valuable starting materials. This improved atom economy means that less raw material is required to produce the same amount of final product, leading to substantial cost savings in the bill of materials. Additionally, the use of common, commodity-grade solvents like ethanol and THF reduces procurement complexity and cost volatility compared to specialized or highly regulated solvents.
- Enhanced Supply Chain Reliability: The reliance on readily available commercial starting materials, such as methyl 2-bromo-4-fluoro-5-nitrobenzoate, ensures that the supply chain is not vulnerable to bottlenecks associated with custom-synthesized complex intermediates. The robustness of the catalytic hydrogenation step, which operates under mild conditions, reduces the likelihood of batch failures due to thermal runaways or equipment malfunctions. This process stability allows for predictable production scheduling and shorter lead times for order fulfillment. By diversifying the supplier base with partners capable of executing this safer and more efficient route, pharmaceutical companies can build a more resilient supply network that is less susceptible to regulatory shutdowns caused by environmental or safety violations.
- Scalability and Environmental Compliance: The green chemistry attributes of this process, including the reduction of heavy metal waste and the avoidance of toxic isocyanates, simplify the regulatory approval process for manufacturing sites. Facilities can operate with lower environmental compliance overheads, as the wastewater treatment load is significantly decreased by removing phosphorus and manganese contaminants. This environmental friendliness future-proofs the manufacturing asset against tightening global environmental regulations, ensuring long-term operational continuity. The simplicity of the workup procedures, often involving straightforward filtration and crystallization rather than complex chromatographic separations, makes the technology highly amenable to scaling from kilogram to multi-ton production volumes with consistent quality.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Ripretinib synthesis technology. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, providing clarity on how this method outperforms historical precedents. Understanding these nuances is vital for technical teams evaluating process transfer feasibility and for commercial teams negotiating supply agreements based on cost and quality metrics.
Q: How does the new synthesis route improve safety compared to conventional methods?
A: The novel process replaces the highly toxic and pungent phenyl isocyanate with the safer combination of aniline and triphosgene, significantly reducing occupational health hazards and environmental risks associated with volatile isocyanates.
Q: What are the yield and purity advantages of this patented method?
A: The optimized pathway demonstrates exceptional efficiency, achieving yields up to 98% in early steps and maintaining final product purity above 99.6%, which minimizes downstream purification costs and material waste.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the method utilizes readily available raw materials and avoids complex, multi-step sequences found in prior art, featuring simple operational conditions like ambient pressure hydrogenation that facilitate easy commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ripretinib Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing infrastructure. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising yields and purity specifications demonstrated in patent CN114213411B can be reliably replicated at an industrial level. We maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to monitor every critical process parameter, guaranteeing that every batch of Ripretinib intermediate meets the stringent purity specifications required for oncology applications. Our commitment to quality assurance ensures that the impurity profiles remain well within acceptable limits, safeguarding the efficacy and safety of the final drug product.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced synthesis technology for their supply chains. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this greener, more efficient route for your specific volume requirements. We encourage you to contact us today to discuss your project needs,索取 specific COA data from our recent pilot batches, and review our detailed route feasibility assessments. Together, we can optimize your Ripretinib supply strategy, ensuring cost-effective, sustainable, and uninterrupted access to this life-saving medication for patients worldwide.