Advanced One-Step Synthesis of Pyrrolo[1,2-f][1,2,4]triazine Nucleoside Intermediates for Commercial API Production
The pharmaceutical industry's relentless pursuit of potent antiviral therapeutics has placed a premium on efficient synthetic routes for nucleoside analogs, particularly those mimicking the structure of Remdesivir. Patent CN111574523A introduces a groundbreaking methodology for preparing 1'-substituted carbon nucleoside analog intermediates, specifically targeting the pyrrolo[1,2-f][1,2,4]triazine scaffold which is critical for broad-spectrum antiviral activity. This innovation represents a paradigm shift from multi-step halogenation protocols to a streamlined, one-step direct coupling strategy that significantly enhances process efficiency. By utilizing pyrrolo[1,2-f][1,2,4]triazine-4-amine under chlorosilane protection in an alkaline environment, the invention enables direct reaction with substituted butyrolactones to form the crucial C-C glycosidic bond. This technical breakthrough not only shortens the overall synthetic timeline but also drastically reduces the reliance on hazardous halogen-containing reagents, addressing both economic and environmental concerns for modern reliable pharmaceutical intermediate supplier networks seeking sustainable manufacturing solutions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of riboside compounds based on the pyrrolo[1,2-f][1,2,4]triazine-4-amine core has been plagued by inefficient multi-step sequences that rely heavily on halogenated precursors. As documented in prior art such as CN102596979A and CN105899216A, conventional routes necessitate the initial conversion of the parent amine into 7-bromo or 7-iodo derivatives before coupling with sugar lactones can occur. This prerequisite halogenation step introduces significant operational complexity, requiring additional reagents, extended reaction times, and rigorous purification protocols to remove halogenated impurities. Furthermore, the generation of stoichiometric amounts of halogen waste creates substantial environmental liabilities and increases disposal costs, which directly impacts the cost reduction in API manufacturing initiatives of large-scale producers. The cumulative effect of these inefficiencies results in lower overall yields and higher production costs, making the supply of these critical antiviral intermediates vulnerable to market fluctuations and raw material shortages.
The Novel Approach
In stark contrast to the cumbersome legacy methods, the technology disclosed in CN111574523A employs a direct lithiation strategy that bypasses the need for pre-functionalization of the heterocyclic base. By protecting the amine functionality with chlorosilanes and utilizing strong organolithium bases, the process achieves regioselective activation at the C7 position in situ. This allows for the immediate nucleophilic attack on the carbonyl group of substituted butyrolactones, effectively merging the activation and coupling stages into a single operational sequence. The elimination of the halogenation step not only simplifies the workflow but also improves the atom economy of the process, leading to substantial cost savings and a cleaner reaction profile. This novel approach demonstrates wide substrate applicability, accommodating various substituents on the butyrolactone ring, which is essential for the commercial scale-up of complex pharmaceutical intermediates required for next-generation antiviral drug pipelines.
Mechanistic Insights into Chlorosilane-Mediated Direct Lithiation
The core of this synthetic innovation lies in the precise manipulation of the electronic properties of the pyrrolo[1,2-f][1,2,4]triazine system through temporary silyl protection. The reaction initiates with the deprotonation of the exocyclic amine using a strong base such as sodium hydride (NaH), followed by protection with chlorosilanes like 1,2-bis(chlorodimethylsilyl)ethane or trimethylchlorosilane. This protection serves a dual purpose: it prevents unwanted side reactions at the nitrogen center and potentially enhances the acidity of the adjacent C7 proton, facilitating subsequent metalation. Upon cooling the reaction mixture to cryogenic temperatures, typically below -20°C, a lithium alkyl derivative such as n-butyllithium or lithium diisopropylamide (LDA) is introduced to effect regioselective lithiation. The resulting organolithium species is a highly reactive nucleophile that is perfectly poised for the subsequent carbon-carbon bond-forming event.
![Chemical structure of Pyrrolo[1,2-f][1,2,4]triazine-4-amine showing the core heterocyclic base used in the synthesis](/insights/img/pyrrolo-triazine-nucleoside-synthesis-supplier-20260306043314-01.png)
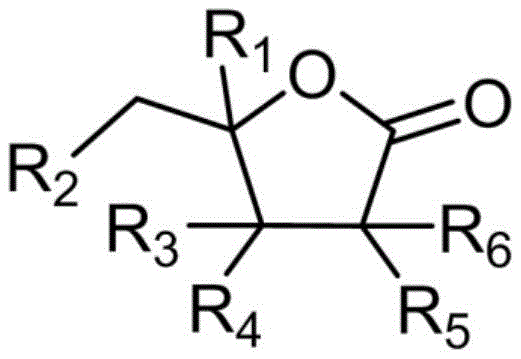
Following the generation of the lithiated intermediate, the reaction proceeds with the addition of the electrophilic partner, the substituted butyrolactone. The general structure of these lactones, as depicted in the patent, allows for significant structural diversity at the R1 through R6 positions, enabling the synthesis of a wide array of non-natural nucleoside analogs. The nucleophilic attack occurs at the carbonyl carbon of the lactone, opening the ring or forming a hemiketal intermediate depending on the specific conditions and workup, ultimately yielding the 1'-substituted carbon nucleoside analog. Maintaining the internal temperature below -60°C during this addition is critical to suppress competing pathways such as over-addition or decomposition of the sensitive organometallic species. This mechanistic precision ensures high-purity nucleoside analogs with minimal impurity profiles, a key requirement for regulatory compliance in pharmaceutical manufacturing.
Impurity control in this process is inherently managed by the selectivity of the lithiation step and the stability provided by the chlorosilane protecting group. In conventional halogenated routes, metal-halogen exchange can sometimes lead to mixtures of regioisomers or homocoupling byproducts. However, the direct lithiation method described here leverages the intrinsic electronic bias of the protected heterocycle to direct the lithium atom specifically to the C7 position. Furthermore, the use of anhydrous conditions and inert atmosphere (nitrogen protection) throughout the procedure prevents hydrolysis of the reactive intermediates. The final quenching step, performed by warming to ≥0°C and adding acidic substances like ammonium chloride or acetic acid, ensures the clean conversion of the alkoxide intermediate to the final alcohol product. This rigorous control over reaction parameters translates to a robust process capable of delivering consistent quality, which is vital for reducing lead time for high-purity pharmaceutical intermediates in a fast-paced drug development environment.
How to Synthesize 1'-Substituted Carbon Nucleoside Analogs Efficiently
The practical execution of this synthesis involves a carefully orchestrated sequence of reagent additions and temperature controls designed to maximize yield and safety. Operators must begin by establishing an inert atmosphere and ensuring all solvents, particularly tetrahydrofuran (THF), are strictly anhydrous to prevent premature quenching of the organolithium reagents. The protection step is typically conducted at 0°C or room temperature, followed by a significant cooldown phase to reach the cryogenic window required for lithiation. The addition of the lactone must be performed slowly to manage the exotherm and maintain the integrity of the reaction mixture. Detailed standard operating procedures regarding stoichiometry, addition rates, and workup protocols are essential for successful technology transfer.
- Protect the pyrrolo[1,2-f][1,2,4]triazine-4-amine base using chlorosilanes (e.g., TMSCl or bis-chlorosilanes) in an alkaline environment with NaH.
- Perform regioselective lithiation at the C7 position using n-butyllithium or LDA at cryogenic temperatures between -20°C and -78°C.
- Add the substituted butyrolactone electrophile to the lithiated intermediate, maintain low temperature, then quench with acid and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers compelling strategic advantages that extend beyond simple technical feasibility. By eliminating the discrete halogenation step, the process removes a significant bottleneck from the production schedule, thereby enhancing the overall agility of the supply chain. The reduction in unit operations means fewer opportunities for yield loss and less handling of hazardous materials, which directly correlates to improved operational safety and lower insurance and compliance costs. Furthermore, the starting material, pyrrolo[1,2-f][1,2,4]triazine-4-amine, is more readily accessible and cost-effective than its halogenated counterparts, providing a buffer against raw material price volatility. This structural simplification of the supply chain ensures greater continuity of supply for critical antiviral ingredients.
- Cost Reduction in Manufacturing: The most immediate financial benefit arises from the drastic simplification of the synthetic route. By removing the need for expensive halogenating agents and the associated purification steps to remove halogenated impurities, the overall cost of goods sold (COGS) is significantly lowered. Additionally, the reduction in waste generation, particularly halogenated organic waste, leads to substantial savings in waste disposal and environmental treatment fees. The higher atom economy of the direct coupling method means that a greater proportion of raw materials end up in the final product, further optimizing resource utilization and driving down the effective cost per kilogram of the active intermediate.
- Enhanced Supply Chain Reliability: Relying on non-halogenated starting materials mitigates the risk of supply disruptions often associated with specialized halogenated reagents. The simplified process flow reduces the dependency on multiple vendors for different stages of synthesis, consolidating production into a more manageable and controllable operation. This consolidation allows for better inventory management and faster response times to fluctuating market demands. Moreover, the robustness of the reaction conditions, utilizing common solvents and reagents like n-butyllithium, ensures that production can be easily replicated across different manufacturing sites without the need for highly specialized or scarce equipment, thereby strengthening the resilience of the global supply network.
- Scalability and Environmental Compliance: From a scale-up perspective, the one-pot nature of this reaction minimizes the need for intermediate isolations, which are often the most challenging steps to scale due to filtration and drying bottlenecks. The process is compatible with standard reactor setups equipped for low-temperature operations, making it accessible for existing manufacturing facilities. Environmentally, the avoidance of heavy halogen loads aligns with increasingly stringent global regulations on chemical manufacturing emissions. This 'greener' profile not only reduces the environmental footprint but also facilitates smoother regulatory approvals and audits, positioning the manufacturer as a responsible partner in the sustainable production of life-saving medicines.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis method. These insights are derived directly from the experimental data and claims presented in CN111574523A, providing a factual basis for decision-making. Understanding the nuances of temperature control, reagent selection, and workup procedures is essential for R&D teams evaluating this technology for pilot or commercial scale production. The answers below reflect the specific advantages and operational parameters defined in the intellectual property.
Q: How does this novel method improve upon traditional halogenated routes for nucleoside synthesis?
A: Traditional methods require pre-halogenation (bromination or iodination) of the base, adding extra steps and generating hazardous halogen waste. This patent describes a direct lithiation approach that eliminates the halogenation step entirely, reducing raw material costs and environmental impact while simplifying the workflow.
Q: What are the critical temperature controls required for high yield in this reaction?
A: Precise thermal management is essential. The lithiation step typically requires cooling to below -20°C, often down to -78°C, to prevent side reactions. The subsequent addition of the lactone also benefits from cryogenic conditions (-60°C to -78°C) to ensure regioselectivity and minimize decomposition of the reactive organolithium species.
Q: Is this process scalable for industrial production of antiviral intermediates?
A: Yes, the process uses standard reagents like n-butyllithium and common solvents like THF. While it requires low-temperature capabilities, the elimination of multiple isolation steps and the use of robust chlorosilane protection make it highly suitable for kilogram-to-ton scale manufacturing of complex nucleoside analogs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1'-Substituted Carbon Nucleoside Analog Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the race to develop new antiviral therapies. Our team of expert chemists has thoroughly analyzed the methodology described in CN111574523A and is fully prepared to leverage this technology for your specific project needs. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. Our state-of-the-art facilities are equipped with the necessary cryogenic capabilities and rigorous QC labs to meet stringent purity specifications, guaranteeing that every batch of nucleoside intermediate meets the highest standards of quality and consistency required by global regulatory bodies.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs through the adoption of this advanced synthesis platform. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in complex organic synthesis can accelerate your drug development timeline while maximizing your return on investment.