Advanced One-Pot Synthesis of Z-Configuration Alkenyl Ester Triazoles for Commercial Scale-Up
Advanced One-Pot Synthesis of Z-Configuration Alkenyl Ester Triazoles for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking more efficient pathways to construct complex heterocyclic scaffolds, particularly those exhibiting potent biological activities. Patent CN108997233B introduces a groundbreaking synthesis method for (Z)-configuration alkenyl ester triazole compounds, addressing critical bottlenecks in traditional organic synthesis. This innovation utilizes a Lewis acid promoted tandem reaction sequence that seamlessly integrates [3+2]-cycloaddition, furan ring opening, and esterification into a single operational step. By leveraging simple and readily available starting materials such as alkyl or aryl azides, various alcohols, and substituted 5-halogenated-2-furfuryl alcohol compounds, this method achieves high step economy and product structure diversity. For R&D directors and process chemists, this represents a significant leap forward in constructing the conjugated skeleton structure of 1,2,3-triazole and alpha,beta-unsaturated esters, which are pivotal motifs in the development of new anticancer agents and other bioactive molecules.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of complex alkenyl ester triazole structures has been plagued by inefficient multistep organic synthesis processes that demand rigorous separation and purification protocols at every stage. Traditional routes often rely on copper-catalyzed Huisgen azide-alkyne cycloadditions or reactions involving alkynes with specific tonicity, which frequently suffer from limited substrate scope and harsh reaction conditions. These conventional methods typically lack the synthetic capacity to efficiently generate the specific conjugated systems required for advanced drug discovery without incurring substantial material loss. Furthermore, the necessity to isolate unstable intermediates between steps not only drives up operational costs but also increases the risk of decomposition, leading to overall lower yields and compromised purity profiles that are unacceptable for high-grade pharmaceutical applications.
The Novel Approach
In stark contrast, the novel approach detailed in the patent employs a sophisticated one-pot tandem reaction strategy that drastically simplifies the synthetic landscape. By utilizing a low-cost Lewis acid catalyst, specifically tin(IV) chloride (SnCl4), in the presence of a tertiary amine base, the reaction orchestrates a complex cascade of transformations under mild conditions ranging from -20°C to room temperature. This methodology allows for the direct conversion of simple precursors into highly functionalized (Z)-configuration alkenyl ester triazole compounds with remarkable stereospecificity. The ability to bypass intermediate isolation steps not only enhances atom utilization but also significantly reduces the generation of chemical waste, aligning perfectly with modern green chemistry principles while delivering superior structural diversity for library synthesis.
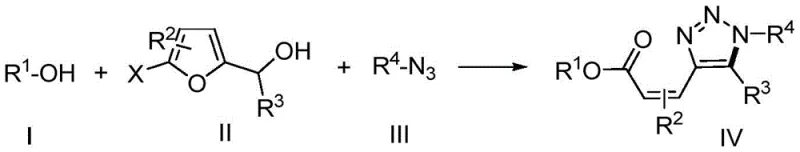
Mechanistic Insights into SnCl4-Catalyzed Tandem Cyclization
The core of this transformative synthesis lies in the precise mechanistic role played by the Lewis acid catalyst, SnCl4, which acts as a powerful activator for the furan ring system. Upon coordination with the oxygen atom of the 5-halogenated-2-furfuryl alcohol, the Lewis acid increases the electrophilicity of the furan ring, facilitating the initial nucleophilic attack by the azide species. This triggers the [3+2]-cycloaddition event, forming a transient triazoline intermediate that is primed for the subsequent ring-opening step. The presence of the tertiary amine is crucial here, as it assists in the deprotonation and stabilization of intermediates, ensuring the smooth progression of the cascade towards the final esterified product without stalling at the cycloaddition stage.
Furthermore, the mechanism inherently controls the stereochemical outcome of the reaction, favoring the formation of the (Z)-configuration isomer with high fidelity. As the furan ring opens and the esterification occurs concurrently with the alcohol substrate, the conjugated system forms in a geometry that minimizes steric hindrance between the bulky triazole ring and the ester group. This stereospecificity is vital for pharmaceutical applications where the biological activity is often strictly dependent on the geometric configuration of the double bond. The robust nature of this catalytic cycle ensures that even with diverse substrates, including sterically hindered cycloalkyl azides or complex chiral alcohols like cholesterol, the reaction maintains its efficiency and selectivity, providing a reliable platform for generating high-purity intermediates.
How to Synthesize (Z)-Alkenyl Ester Triazole Efficiently
Executing this synthesis requires careful attention to reagent stoichiometry and temperature control to maximize the yield of the desired (Z)-isomer. The process begins with the preparation of a dry reaction environment, typically using dichloromethane as the solvent to ensure solubility of all three components: the alcohol, the 5-halo-2-furfuryl alcohol, and the azide. The addition of the Lewis acid catalyst must be performed at low temperatures, specifically around -20°C, to manage the exothermic nature of the activation and prevent side reactions. Following the addition of the tertiary amine base, the reaction mixture is allowed to warm slowly to room temperature, a critical phase where the tandem cyclization and ring-opening events take place. Detailed standardized synthetic steps for optimizing this protocol are provided in the guide below.
- Prepare the reaction mixture by combining 5-halogenated-2-furfuryl alcohol, alkyl or aryl azide, and the desired alcohol substrate in dried dichloromethane under inert atmosphere.
- Cool the solution to -20°C and sequentially add the Lewis acid catalyst SnCl4 and a tertiary amine base such as triethylamine or pyridine.
- Allow the reaction to warm slowly to room temperature, monitor progress via TLC, and upon completion, quench with saturated sodium bicarbonate followed by standard extraction and purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis method offers profound strategic advantages that extend beyond mere technical feasibility. The consolidation of multiple synthetic steps into a single pot operation fundamentally alters the cost structure of manufacturing these valuable intermediates. By eliminating the need for intermediate isolation, filtration, and drying between steps, the process drastically reduces the consumption of solvents and consumables, leading to substantial cost savings in raw material procurement. Additionally, the use of inexpensive and widely available reagents like SnCl4 and common alcohols ensures that the supply chain remains resilient against market fluctuations, providing a stable foundation for long-term production planning.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts like copper, which often require expensive removal processes to meet pharmaceutical purity standards, results in a streamlined downstream processing workflow. This reduction in purification complexity translates directly into lower operational expenditures and shorter batch cycle times. Furthermore, the high atom economy of the tandem reaction means that a greater proportion of the starting mass is converted into the final product, minimizing waste disposal costs and maximizing the return on investment for every kilogram of raw material purchased.
- Enhanced Supply Chain Reliability: The broad substrate scope of this reaction allows manufacturers to utilize a wide variety of commercially sourced alcohols and azides, reducing dependency on single-source specialty chemicals. This flexibility enables rapid adaptation to supply disruptions by allowing for the substitution of structurally similar reagents without compromising the integrity of the final product. The robustness of the reaction conditions, which do not require extreme pressures or cryogenic temperatures beyond standard cooling, further ensures that production can be maintained consistently across different manufacturing sites with varying equipment capabilities.
- Scalability and Environmental Compliance: From an environmental compliance perspective, the reduction in solvent usage and waste generation aligns with increasingly stringent global regulations on chemical manufacturing emissions. The simplicity of the workup procedure, involving standard aqueous quenching and extraction, facilitates easier scale-up from laboratory to pilot and commercial production scales. This scalability ensures that as demand for these triazole intermediates grows in the oncology sector, the supply chain can expand capacity rapidly without the need for complex new infrastructure or specialized hazardous waste treatment facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on how this method compares to existing industry standards. Understanding these details is essential for making informed decisions about integrating this technology into your current manufacturing portfolio.
Q: What represents the primary advantage of this tandem reaction over traditional multistep synthesis?
A: The primary advantage lies in the exceptional step economy, where cycloaddition, ring-opening, and esterification occur in a single pot, eliminating intermediate isolation and significantly reducing waste and processing time.
Q: How does the method ensure high stereoselectivity for the (Z)-configuration?
A: The Lewis acid promoted mechanism inherently favors the formation of the thermodynamically stable (Z)-isomer during the furan ring-opening and subsequent conjugation with the ester group, ensuring high stereospecificity without additional isomer separation.
Q: Is this synthesis method scalable for industrial production of API intermediates?
A: Yes, the use of commercially available reagents like SnCl4 and simple alcohols, combined with mild reaction conditions ranging from -20°C to room temperature, makes the process highly amenable to large-scale commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alkenyl Ester Triazole Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this Lewis acid catalyzed tandem reaction for the production of high-value pharmaceutical intermediates. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from benchtop discovery to full-scale manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of (Z)-alkenyl ester triazole meets the exacting standards required for clinical and commercial drug development.
We invite you to collaborate with our technical team to explore how this innovative synthesis route can optimize your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits tailored to your volume needs. We encourage you to contact our technical procurement team today to obtain specific COA data and comprehensive route feasibility assessments, ensuring that your supply chain is built on a foundation of scientific excellence and commercial reliability.