Advanced Manufacturing of Velpatasvir Intermediates: A Technical Breakthrough for Commercial Scale-Up
The global demand for direct-acting antiviral agents, particularly for Hepatitis C treatment, has necessitated the development of robust and scalable synthetic routes for key active pharmaceutical ingredients like Velpatasvir (GS5816). Patent CN107759577B, published in March 2020, introduces a transformative preparation method for GS5816 intermediates that directly addresses the critical bottlenecks of chiral control and purification complexity found in earlier generations of synthesis. This technical disclosure outlines a multi-step pathway that achieves exceptional chemical and optical purity exceeding 99.50%, with all individual impurities maintained below 0.10%, thereby meeting the rigorous standards required for active pharmaceutical ingredient (API) manufacturing. By leveraging mild reaction conditions and avoiding specialized purification equipment, this methodology offers a compelling value proposition for reliable pharmaceutical intermediate suppliers seeking to optimize their production lines for high-volume commercial output.
For procurement managers and supply chain heads, the implications of this patent extend far beyond mere chemical novelty; it represents a fundamental shift towards cost reduction in pharmaceutical intermediate manufacturing. Traditional methods for synthesizing complex heterocyclic structures often rely heavily on column chromatography for purification, a technique that is notoriously difficult to scale, consumes vast quantities of silica and solvents, and introduces significant variability in batch-to-batch consistency. The novel approach detailed in this patent circumvents these issues by designing a synthetic route where intermediates can be isolated through simple filtration and washing processes. This operational simplicity translates directly into enhanced supply chain reliability, as the dependency on specialized chromatographic columns and the associated downtime for column packing and regeneration is completely eliminated, ensuring a more continuous and predictable production flow for critical hepatitis C therapeutics.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methods for the synthesis of Velpatasvir and its precursors have historically struggled with the delicate balance between reaction efficiency and stereochemical integrity. The background section of the patent highlights that existing technologies often suffer from weak fluorescence of raw materials, making process monitoring difficult, and more critically, they face substantial challenges in controlling chirality during the synthesis process. When chiral isomers are not strictly controlled, the resulting intermediate possesses poor optical purity, which necessitates extensive and costly downstream purification efforts to meet regulatory specifications. Furthermore, the reliance on column chromatography for isolating intermediates not only inflates the cost of goods sold (COGS) due to material waste but also creates a bottleneck in commercial scale-up of complex pharmaceutical intermediates, as scaling chromatographic processes requires disproportionate increases in equipment footprint and solvent handling capacity compared to crystallization-based workflows.
The Novel Approach
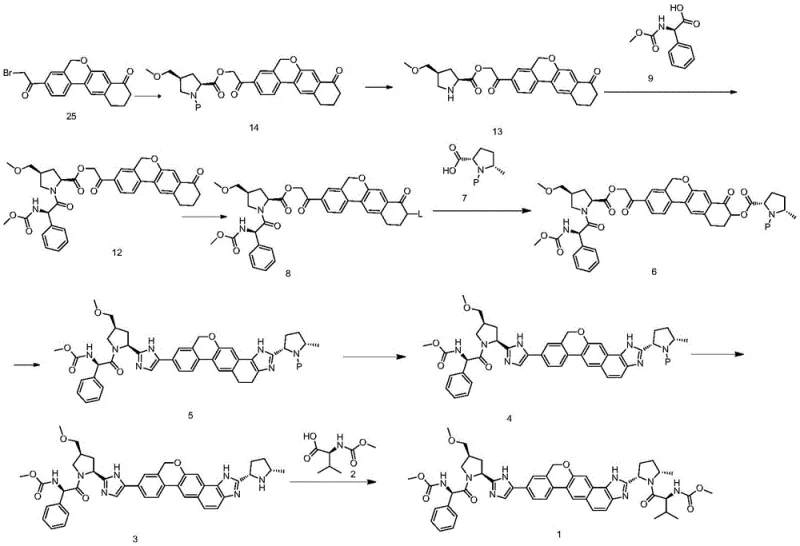
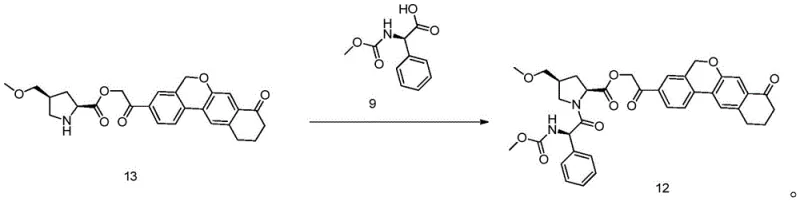
The innovative strategy presented in this patent overcomes these historical deficiencies by introducing a sequence of reactions that are inherently cleaner and more selective. Central to this approach is the use of specific condensing agents, such as propylphosphonic anhydride (T3P), in amide solvents like DMF, which facilitate high-yield coupling reactions while generating byproducts that are easily removed during aqueous work-ups. The patent describes a method where Compound 13 and Compound 9 undergo condensation to form Compound 12 under mild temperatures ranging from 0 to 40°C, a condition that preserves the stereochemical configuration of the sensitive chiral centers. By optimizing molar ratios and utilizing precise temperature controls, the process ensures that the reaction proceeds to completion with minimal formation of side products, allowing the final product to be isolated simply by cooling, adding water, and filtering. This shift from complex separation science to straightforward physical isolation marks a significant advancement in process chemistry, enabling manufacturers to achieve high purity levels without the need for special purification equipment.
Mechanistic Insights into T3P-Mediated Condensation and Cyclization
A deep dive into the reaction mechanisms reveals why this specific pathway offers superior control over impurity profiles. The formation of the key amide bond between Compound 13 and Compound 9 is mediated by T3P, which activates the carboxylic acid group of Compound 9 to form a highly reactive mixed anhydride species. This activation mode is particularly advantageous because the resulting phosphoric acid byproducts are water-soluble, allowing them to be effortlessly washed away during the post-reaction work-up, thus preventing contamination of the final intermediate. The patent specifies that the molar ratio of the condensing agent to the substrate is carefully tuned, preferably between 1:1 and 1:1.5, to ensure complete conversion while minimizing excess reagent waste. This precision in stoichiometry, combined with the use of inert protective gases like nitrogen or argon, creates an environment where oxidative degradation and hydrolysis are suppressed, leading to the reported yields of up to 97.0% for Compound 12 with HPLC purity exceeding 98.71%.
Furthermore, the subsequent cyclization steps to form the imidazole core, such as the conversion of Compound 12 to Compound 17 using ammonium acetate, demonstrate a sophisticated understanding of heterocyclic chemistry. The use of ammonium salts in aromatic hydrocarbon solvents like toluene at elevated temperatures (80-100°C) promotes the dehydration and cyclization necessary to close the ring structure without compromising the adjacent chiral centers. The patent notes that this condensation can be monitored effectively via TLC or HPLC, with the disappearance of the starting material serving as a clear endpoint. The ability to drive these equilibrium-limited reactions to completion through the removal of water or the use of excess ammonium salt ensures that the final API precursor possesses the requisite structural integrity. This mechanistic robustness is crucial for maintaining the stringent purity specifications required for antiviral drugs, where even trace impurities can impact therapeutic efficacy or safety profiles.
How to Synthesize Velpatasvir Intermediate Efficiently
The practical implementation of this synthesis route involves a series of well-defined unit operations that are familiar to process chemists but optimized for maximum efficiency. The process begins with the preparation of the amine component, followed by the critical coupling step and subsequent heterocyclic ring formations. Each stage is designed to maximize yield while minimizing the generation of hazardous waste, aligning with modern green chemistry principles. The detailed procedural steps involve specific solvent choices, such as switching from DMF for coupling to dichloromethane for halogenation, which optimizes solubility and reaction kinetics at each stage. For a comprehensive guide on executing these transformations with precision, please refer to the standardized protocol outlined below.
- Prepare Compound 13 by removing the amino protecting group from Compound 14 using acid in an ester solvent like isopropyl acetate.
- Conduct a condensation reaction between Compound 13 and Compound 9 in DMF using a propylphosphonic anhydride (T3P) coupling agent.
- Isolate Compound 12 via filtration and washing after the reaction completes, achieving high purity without column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this patented methodology offers substantial strategic advantages for organizations managing the supply of hepatitis C therapeutics. The primary benefit lies in the drastic simplification of the downstream processing workflow. By eliminating the need for column chromatography, manufacturers can significantly reduce the consumption of silica gel and organic solvents, which are major cost drivers in fine chemical production. This reduction in material usage not only lowers the direct variable costs but also diminishes the environmental burden associated with solvent disposal, facilitating easier compliance with increasingly strict environmental regulations. Moreover, the simplified work-up procedure, which relies on filtration and washing, allows for faster batch turnover times, thereby increasing the overall throughput of the production facility without requiring capital investment in new equipment.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, primarily driven by the removal of chromatographic purification steps. In traditional synthesis, chromatography can account for a significant portion of the total manufacturing cost due to the high price of stationary phases and the large volumes of solvents required for elution. By designing a route where intermediates precipitate or can be extracted cleanly, the process eliminates these expenses entirely. Additionally, the high yields reported in the patent examples, often exceeding 95% for key steps, mean that less starting material is required to produce the same amount of final product, further driving down the cost of raw materials. This efficiency makes the production of high-purity pharmaceutical intermediates much more economically viable, allowing suppliers to offer competitive pricing while maintaining healthy margins.
- Enhanced Supply Chain Reliability: Supply chain resilience is heavily dependent on the predictability of manufacturing processes. The mild reaction conditions described in the patent, typically ranging from 0°C to 40°C for coupling and up to 100°C for cyclization, are easily achievable with standard industrial reactors, reducing the risk of thermal runaways or equipment failures. The use of commercially available reagents like T3P, ammonium acetate, and common solvents ensures that the supply of raw materials is stable and not subject to the volatility of niche specialty chemicals. This accessibility of inputs, combined with the robustness of the reaction conditions, minimizes the risk of production delays caused by reagent shortages or process deviations, ensuring a steady flow of intermediates to downstream API manufacturers.
- Scalability and Environmental Compliance: Scaling chemical processes from the laboratory to the plant floor often introduces unforeseen challenges, particularly regarding heat transfer and mixing efficiency. However, the heterogeneous nature of some work-up steps in this process, such as the precipitation of products upon water addition, actually aids in scalability by simplifying isolation. The avoidance of column chromatography removes a major scale-up barrier, as industrial-scale chromatography is complex and expensive to operate. Furthermore, the reduced solvent load and the ability to recycle certain mother liquors contribute to a smaller environmental footprint. This alignment with green chemistry principles not only satisfies regulatory requirements but also appeals to end-clients who are increasingly prioritizing sustainability in their supplier selection criteria, enhancing the marketability of the produced intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and benefits of the technology. Understanding these details is essential for technical teams evaluating the potential for technology transfer or licensing.
Q: How does this patent address chiral purity challenges in Velpatasvir synthesis?
A: The patent utilizes mild reaction conditions and specific coupling agents like T3P that minimize racemization. Furthermore, the process avoids harsh purification steps like column chromatography, relying instead on crystallization which inherently enhances optical purity to greater than 99.50%.
Q: What are the primary cost drivers eliminated in this new manufacturing route?
A: The most significant cost reduction comes from eliminating column chromatography, which is expensive and difficult to scale. Additionally, the use of commercially available reagents and simplified work-up procedures (filtration and washing) drastically reduces solvent consumption and processing time.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method is suitable for industrial production. The reactions operate under mild temperatures (often 0-40°C), use standard solvents like DMF and DCM, and produce intermediates with impurities less than 0.10%, meeting strict API standards required for commercial supply chains.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Velpatasvir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of having a robust and scalable supply chain for life-saving antiviral medications. Our team of expert process chemists has thoroughly analyzed the technological advancements presented in patent CN107759577B and is fully equipped to translate these laboratory successes into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high purity and yield demonstrated in the patent are maintained at an industrial level. Our state-of-the-art facilities are designed to handle the specific solvent systems and reaction conditions required for this synthesis, including the safe handling of coupling agents and the precise temperature control needed for chiral integrity. With our stringent purity specifications and rigorous QC labs, we guarantee that every batch of Velpatasvir intermediate meets the highest international standards for quality and safety.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to optimize their supply chains for Hepatitis C treatments. By leveraging our expertise in process optimization, we can help you achieve significant efficiencies and cost savings. We encourage you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our team is ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term production goals and ensure the uninterrupted availability of high-quality intermediates for the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →