Enantioselective Synthesis of Alpha-Fluoro Phosphine Oxides for Advanced Pharmaceutical Manufacturing
Introduction to Advanced Enantioselective Fluorination Technology
The landscape of organic synthesis for pharmaceutical intermediates is constantly evolving, driven by the demand for higher purity and specific stereochemical configurations. A significant breakthrough in this domain is documented in Chinese Patent CN112142791A, which discloses a novel method for the synthesis of α-fluoro-β-carbonyl-diarylphosphine oxide compounds with enantioselective diversity. This technology addresses a critical gap in the field of organophosphorus chemistry, where the introduction of a fluorine atom at the α-position of a carbonyl group adjacent to a phosphine oxide moiety has historically been challenging due to the complexity of fluorine substitution and the difficulty in controlling stereochemistry. The patent outlines a robust protocol that utilizes β-carbonyl-diarylphosphine oxide compounds as substrates, reacting them with fluorinating reagents in the presence of chiral catalysts and alkaline solutions to yield α-asymmetric-fluorine substituted products with exceptional optical purity.
This innovation is particularly relevant for the development of high-purity pharmaceutical intermediates, as these fluorinated phosphine oxides serve as versatile synthons for the Horner-Wadsworth-Emmons (HWE) reaction and can be transformed into various bioactive molecules. By leveraging cheap and commercially available cinchona alkaloids and their derivatives as chiral catalysts, the process achieves economic efficiency without compromising on the quality of the final product. The ability to economically and efficiently realize asymmetric α-fluorination with high synthesis yields makes this method highly suitable for industrial production, offering a reliable pathway for manufacturers seeking to optimize their supply chains for complex organofluorine compounds.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this technological advancement, the halogenation of phosphonates, particularly at the α-position, was a subject of intense research but suffered from significant limitations regarding selectivity and scope. Historical literature indicates that while α-chlorination and α-bromination reactions were achievable, the corresponding fluorination reactions were fraught with difficulties. Existing methods often relied on electrochemical approaches or specific Lewis acid catalysts that yielded products with low purity and poor enantioselectivity. For instance, previous attempts to synthesize α-fluoro phosphonates using sodium cyanide or other reagents resulted in extremely low yields, while asymmetric chlorination methods using chiral spiropyridine monooxazoline ligands achieved only modest enantiomeric excess values, with the highest reported ee values reaching merely 35%. Furthermore, the small atomic radius and high electronegativity of fluorine make the asymmetric fluorination process inherently more complex than its chloro- or bromo-counterparts, often leading to racemic mixtures that require costly and inefficient resolution steps.
The Novel Approach
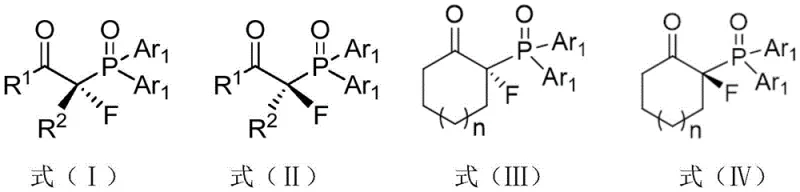
In stark contrast to these conventional limitations, the method described in patent CN112142791A introduces a paradigm shift by employing organocatalysis to drive the enantioselective diversity of the reaction. By utilizing β-carbonyl-diarylphosphine oxide compounds represented by general formulas (V) or (VI) as starting materials, the process enables the direct introduction of a fluorine atom with precise stereochemical control. The novelty lies in the specific combination of substrates with fluorinating reagents such as NFSI, Selectfluor, or Selectfluor II, mediated by a suite of accessible chiral catalysts including quinine, quinidine, cinchonine, and their hydrogenated derivatives. This approach not only overcomes the barrier of low enantioselectivity found in prior art but also simplifies the reaction process, making it safe and easy to operate. The result is the production of single-crystal形态的 α-fluoro-β-carbonyl-diarylphosphine oxide compounds with ee values consistently exceeding 90% and reaching up to 99%, representing a substantial improvement in product quality and process reliability for the fine chemical industry.
Mechanistic Insights into Cinchona Alkaloid-Catalyzed Asymmetric Fluorination
The core of this synthetic breakthrough lies in the sophisticated interaction between the chiral organocatalyst and the substrate during the fluorination event. The mechanism likely involves the activation of the β-carbonyl-diarylphosphine oxide substrate by the basic environment provided by additives such as potassium carbonate, cesium carbonate, or organic bases like DBU and DMAP derivatives. The chiral cinchona alkaloid catalyst creates a sterically defined environment around the reactive enolate intermediate, directing the approach of the electrophilic fluorine source to one specific face of the planar intermediate. This facial selectivity is crucial for establishing the new stereocenter at the α-position with high fidelity. The use of DMAP derivatives, as illustrated in the patent's structural examples, further suggests that nucleophilic catalysis or hydrogen-bonding networks may play a pivotal role in stabilizing the transition state, thereby lowering the activation energy for the desired enantiomer while suppressing the formation of the undesired mirror image.
Furthermore, the impurity control mechanism is intrinsically linked to the high enantioselectivity of the catalytic system. By ensuring that the reaction proceeds predominantly through a single stereochemical pathway, the formation of diastereomeric impurities and racemic byproducts is minimized. The patent data demonstrates that varying the substituents on the aromatic rings (R1, R2, and Ar1) does not significantly compromise the stereocontrol, indicating a robust catalytic cycle that tolerates electronic and steric variations. This mechanistic robustness is essential for R&D directors who require consistent quality across different batches and analogues. The ability to obtain single crystals of the product, as confirmed by X-ray diffraction in the patent examples, provides definitive proof of the absolute configuration, ensuring that the synthesized intermediates meet the stringent purity specifications required for downstream pharmaceutical applications.
How to Synthesize Alpha-Fluoro-Beta-Carbonyl-Diarylphosphine Oxides Efficiently
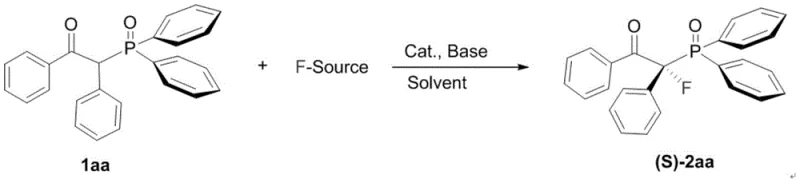
To implement this cutting-edge synthesis in a laboratory or pilot plant setting, operators must adhere to the optimized conditions detailed in the patent to maximize yield and enantiomeric excess. The general procedure involves dissolving the β-carbonyl-diarylphosphine oxide substrate in an organic solvent such as acetonitrile, dichloromethane, or acetone, followed by the sequential addition of the chiral catalyst and the base. The reaction is typically conducted at low temperatures, ranging from -10°C to 0°C, over a period of approximately 72 hours to ensure complete conversion and high stereoselectivity. Detailed standardized synthesis steps for specific analogues are provided in the guide below, which serves as a foundational reference for scaling up the process.
- Prepare the reaction mixture by combining the beta-carbonyl-diarylphosphine oxide substrate with a chiral cinchona alkaloid catalyst and an organic base in a suitable solvent such as acetonitrile or dichloromethane.
- Add the electrophilic fluorinating reagent, such as NFSI or Selectfluor, to the reaction vessel while maintaining a controlled low temperature typically between -10°C and 0°C to ensure high stereoselectivity.
- Stir the reaction for approximately 72 hours until completion, then quench with saturated sodium thiosulfate, extract with organic solvents, and purify via column chromatography to isolate the high-purity chiral product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible strategic benefits that extend beyond mere technical performance. The primary advantage is the significant cost reduction in fine chemical manufacturing achieved through the elimination of expensive transition metal catalysts and the use of readily available, commodity-grade organocatalysts. Since the catalysts employed, such as cinchona alkaloids, are derived from natural sources and are produced on a massive industrial scale, their price volatility is low, and their supply is secure. This stability translates directly into a more predictable cost structure for the final API intermediates, allowing companies to better forecast their raw material expenses and maintain healthy profit margins even in fluctuating market conditions.
- Cost Reduction in Manufacturing: The process eliminates the need for precious metal catalysts and complex ligand systems that often characterize asymmetric synthesis, thereby removing the associated costs of metal removal and purification steps. By relying on organocatalysis, the downstream processing is simplified, reducing solvent consumption and waste treatment costs. This streamlined workflow ensures that the overall production cost is substantially lower compared to traditional methods that require rigorous purification to remove metal residues, making the final product more competitive in the global marketplace.
- Enhanced Supply Chain Reliability: The reliance on commercially available reagents and solvents means that the supply chain is less vulnerable to disruptions caused by the scarcity of specialized chemicals. Since the key catalysts and fluorinating agents are standard items in the chemical inventory of most suppliers, lead times for raw material procurement are significantly reduced. This availability ensures continuous production schedules and minimizes the risk of delays in delivering high-purity pharmaceutical intermediates to clients, thereby strengthening the reliability of the supply network.
- Scalability and Environmental Compliance: The reaction conditions are mild and operate at near-ambient or slightly sub-ambient temperatures, which reduces energy consumption and enhances safety profiles for large-scale operations. The simplicity of the workup procedure, involving standard aqueous quenching and extraction, facilitates easy scale-up from gram to kilogram scales without requiring specialized equipment. Additionally, the avoidance of heavy metals aligns with increasingly stringent environmental regulations and green chemistry principles, reducing the regulatory burden and disposal costs associated with hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this enantioselective fluorination technology. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the process capabilities and limitations for potential partners and stakeholders.
Q: What represents the primary advantage of this fluorination method over conventional halogenation?
A: Unlike conventional methods that often result in racemic mixtures or are limited to chlorination and bromination with poor enantioselectivity, this patented process utilizes cheap cinchona alkaloid catalysts to achieve enantiomeric excess (ee) values exceeding 90%, often reaching up to 99%.
Q: Which fluorinating reagents are compatible with this synthetic protocol?
A: The protocol demonstrates high compatibility with standard electrophilic fluorinating sources including N-fluorobisbenzenesulfonamide (NFSI), Selectfluor, and Selectfluor II, allowing flexibility in reagent sourcing for industrial scale-up.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the method is explicitly designed for industrial applicability, utilizing commercially available and inexpensive catalysts, simple operational procedures, and standard organic solvents, which facilitates significant cost reduction in fine chemical manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Fluoro-Beta-Carbonyl-Diarylphosphine Oxide Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of the synthetic methodologies outlined in patent CN112142791A for the production of advanced pharmaceutical intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to industrial reality is seamless and efficient. Our commitment to quality is underpinned by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify the enantiomeric excess and chemical purity of every batch, guaranteeing that our clients receive materials that meet the highest global standards.
We invite forward-thinking pharmaceutical and agrochemical companies to collaborate with us to leverage this cost-effective and high-yielding technology for their specific project needs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements, as well as obtain specific COA data and route feasibility assessments for your target molecules. Let us help you optimize your supply chain and accelerate your drug development timeline with our reliable supply of high-performance organofluorine intermediates.