Advanced Manufacturing Of EFCP Voriconazole Intermediate Via Novel Non-Grignard Synthetic Route
The pharmaceutical landscape for antifungal treatments has been significantly transformed by the advent of second-generation triazoles, with Voriconazole standing out as a critical therapeutic agent for invasive aspergillosis and other serious fungal infections. The commercial viability of such high-value Active Pharmaceutical Ingredients (APIs) relies heavily on the efficiency and cost-effectiveness of their key intermediates. Patent CN101671309B introduces a groundbreaking synthesis method for 6-ethyl-4-hydroxyl-5-fluorine-2-cloro pyridine ammonium salt, commonly referred to as EFCP, which serves as a pivotal building block in the Voriconazole supply chain. This innovation addresses long-standing bottlenecks in traditional manufacturing by replacing hazardous and expensive reagents with accessible alternatives, thereby enhancing the overall sustainability of the production process. As a reliable pharmaceutical intermediate supplier, understanding these technological shifts is essential for securing a competitive edge in the global market. The structural relationship between the target intermediate and the final drug underscores the precision required in its synthesis, as depicted in the molecular overview below.
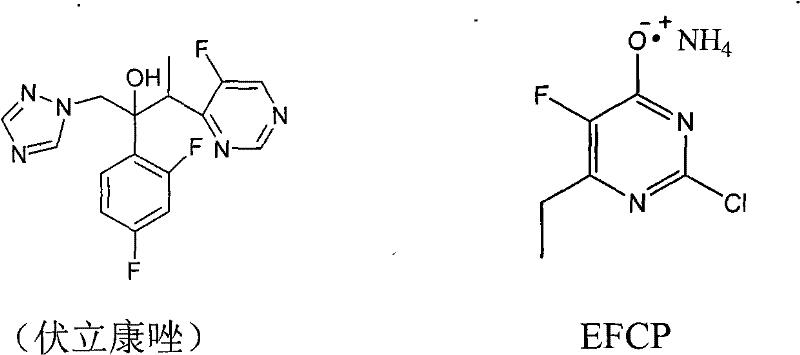
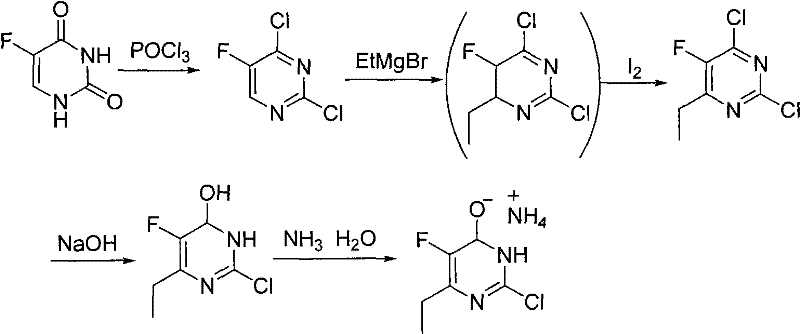
The historical approach to synthesizing EFCP has been fraught with economic and operational challenges that hinder large-scale adoption. Conventional methods typically initiate with 5-fluorouracil, a relatively costly starting material that necessitates a multi-step transformation involving phosphorylation and subsequent Grignard reactions. The reliance on ethylmagnesium bromide introduces severe constraints, requiring strictly anhydrous and oxygen-free environments to prevent catastrophic failure of the reaction, which inflates infrastructure costs. Furthermore, the use of tetrahydrofuran (THF) as a solvent poses significant toxicity and flammability risks, complicating waste management and regulatory compliance. Perhaps most critically, the traditional route depends on iodine as an oxidizing agent, a reagent known for its high price volatility and the generation of substantial iodine-containing waste streams that are difficult to treat. These cumulative factors result in a manufacturing process that is not only capital-intensive but also environmentally burdensome, limiting the ability of producers to offer cost reduction in pharmaceutical intermediate manufacturing to their downstream partners.
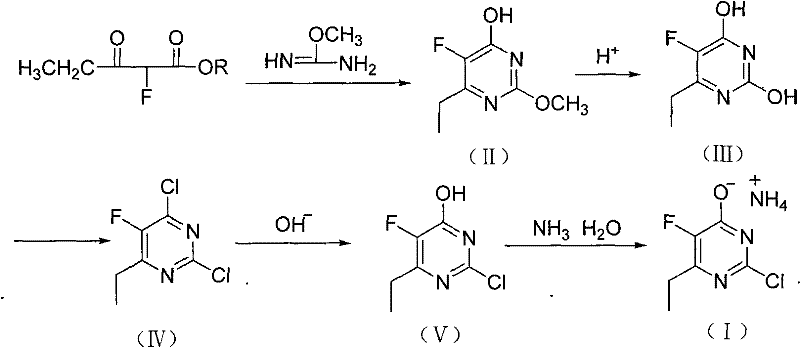
In stark contrast, the novel approach detailed in the patent data presents a streamlined and economically superior pathway that circumvents the pitfalls of the legacy technology. By shifting the starting material to 2-fluoro-3-oxopentanoic acid ester, the new method leverages cheap and easily obtained raw materials that are stable and safe to handle. The core innovation lies in the condensation reaction with O-methylisourea salts, which efficiently constructs the pyrimidine ring without the need for sensitive organometallic reagents. This strategic pivot eliminates the requirement for rigorous moisture exclusion, allowing the reaction to proceed under much milder and more forgiving conditions. The subsequent steps involve standard chlorination and hydrolysis protocols that utilize common industrial reagents like phosphorus oxychloride and sodium hydroxide, further simplifying the operational workflow. This transition represents a paradigm shift towards green chemistry principles, drastically reducing the environmental footprint while simultaneously lowering the barrier to entry for commercial scale-up of complex pharmaceutical intermediates. The comprehensive reaction scheme illustrates the elegance and efficiency of this new synthetic logic.
Mechanistic Insights into the Condensation and Chlorination Cascade
The success of this novel synthesis hinges on the precise control of the condensation reaction between the fluoro-keto ester and the O-methylisourea derivative. In the presence of a sodium alkoxide base, the ester undergoes nucleophilic attack, facilitating the cyclization that forms the pyrimidine core of intermediate (II). The choice of O-methylisourea methyl sulfate salt is particularly advantageous, as it acts as a stable source of the amidine functionality, avoiding the handling difficulties associated with free bases. Following cyclization, the acid-catalyzed hydrolysis step is critical for unmasking the hydroxyl group at the 4-position, setting the stage for the subsequent chlorination. The chlorination mechanism utilizing phosphorus oxychloride (POCl3) is a classic activation strategy where the hydroxyl groups are converted into good leaving groups (chlorides). The addition of N,N-dimethylaniline as an auxiliary reagent likely serves to scavenge the generated HCl and drive the equilibrium forward, ensuring high conversion rates. This sequence demonstrates a robust understanding of heterocyclic chemistry, optimizing each transformation to minimize byproduct formation and maximize the yield of the desired chlorinated scaffold.
Impurity control is inherently superior in this new route due to the absence of competing side reactions typical of Grignard chemistry. In traditional methods, the high reactivity of organomagnesium species often leads to over-addition or reaction with trace moisture, generating difficult-to-remove alcohol impurities. By contrast, the ionic nature of the condensation and hydrolysis steps in the new process allows for straightforward purification via pH adjustment and crystallization. The hydrolysis of intermediate (II) to (III) is conducted under controlled acidic conditions, which selectively cleaves the ether linkage without degrading the sensitive fluorine substituent on the ring. Furthermore, the final ammoniation step to form the salt is performed in an aqueous environment, which naturally washes away organic soluble impurities. This inherent cleanliness of the reaction profile ensures that the final EFCP product meets stringent purity specifications required for API synthesis, reducing the need for extensive downstream polishing and chromatography.
How to Synthesize EFCP Efficiently
The practical implementation of this synthesis requires careful attention to stoichiometry and temperature control, particularly during the initial condensation and the exothermic chlorination phases. The patent outlines a clear progression from the ester starting material through to the final ammonium salt, emphasizing the use of standard laboratory and plant equipment. Operators should note that the molar ratio of the sodium alkoxide to the ester is critical; excessive base can lead to side reactions that diminish yield, while insufficient base results in incomplete conversion. Similarly, the chlorination step benefits from the gradual addition of the catalyst to manage heat evolution effectively. The detailed standardized synthesis steps见下方的指南 provide a granular view of the specific conditions, workup procedures, and isolation techniques necessary to replicate this high-yielding process successfully in a production setting.
- Condense 2-fluoro-3-oxopentanoic acid ester with O-methylisourea salt in sodium alkoxide alcoholic solution to form intermediate (II).
- Perform acid-catalyzed hydrolysis on intermediate (II) to obtain intermediate (III), followed by chlorination with POCl3 to yield compound (IV).
- Execute hydroxyl substitution on compound (IV) using alkaline water solution to get intermediate (V), then ammoniate with ammonia water to form the final salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel EFCP synthesis route offers transformative benefits that extend far beyond simple chemical curiosity. The primary advantage lies in the drastic simplification of the supply chain for raw materials; by eliminating the dependency on imported 5-fluorouracil and volatile iodine, manufacturers can source inputs locally and reliably, insulating the production schedule from global commodity shocks. The removal of the Grignard step also translates to a significant reduction in safety overhead, as the need for specialized inert atmosphere reactors and rigorous drying protocols is obviated. This operational simplification allows for faster batch turnover times and higher throughput in existing facilities without the need for massive capital reinvestment in new infrastructure. Consequently, this leads to substantial cost savings in the overall manufacturing budget, making the final Voriconazole API more competitive in price-sensitive markets.
- Cost Reduction in Manufacturing: The elimination of expensive reagents such as iodine and 5-fluorouracil directly lowers the Bill of Materials (BOM) cost per kilogram of EFCP produced. Furthermore, the avoidance of THF solvent reduces both solvent purchase costs and the expensive waste disposal fees associated with halogenated and ether waste streams. The simplified workup procedures, which rely on basic acid-base extractions rather than complex chromatographic separations, reduce labor hours and consumable usage. These cumulative efficiencies create a leaner production model that enhances profit margins while allowing for more aggressive pricing strategies in the generic pharmaceutical sector.
- Enhanced Supply Chain Reliability: Sourcing 2-fluoro-3-oxopentanoic acid ester is significantly more straightforward than securing high-purity 5-fluorouracil, which is often subject to tight supply constraints due to its use in oncology. The robustness of the new chemical route means that production is less susceptible to delays caused by minor fluctuations in environmental conditions or reagent quality. This reliability ensures a consistent flow of intermediates to API manufacturers, preventing bottlenecks that could halt the production of life-saving antifungal medications. A stable supply of EFCP is crucial for maintaining the continuity of Voriconazole production, safeguarding against market shortages.
- Scalability and Environmental Compliance: The new process is inherently designed for industrial scale-up, utilizing reagents and conditions that are standard in bulk chemical manufacturing. The reduction in toxic waste generation, particularly the absence of iodine-heavy effluents, simplifies compliance with increasingly stringent environmental regulations. This eco-friendly profile not only reduces the risk of regulatory fines but also aligns with the sustainability goals of major multinational pharmaceutical companies. The ability to scale from pilot batches to multi-ton production without encountering the safety hurdles of Grignard chemistry makes this route the preferred choice for long-term commercial partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented EFCP synthesis method. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical implications of switching to this new technology. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams negotiating supply agreements. The answers reflect the consensus on the superiority of the non-Grignard route in terms of both operational safety and economic viability.
Q: Why is the new EFCP synthesis route superior to the conventional 5-fluorouracil method?
A: The new route eliminates the need for expensive 5-fluorouracil, toxic THF solvents, and costly iodine oxidants. It avoids strict anhydrous Grignard conditions, significantly simplifying operations and reducing environmental hazards.
Q: What are the key raw materials for the novel EFCP manufacturing process?
A: The process utilizes 2-fluoro-3-oxopentanoic acid ester as the starting material, reacting with O-methylisourea salts. Subsequent steps involve standard reagents like phosphorus oxychloride and sodium hydroxide, which are readily available and cost-effective.
Q: How does this patent address impurity control in Voriconazole intermediate production?
A: By avoiding complex Grignard reactions and iodine oxidation, the new method reduces side reactions associated with moisture sensitivity and over-oxidation. The simplified purification steps, such as pH adjustment and extraction, ensure high purity suitable for pharmaceutical applications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable EFCP Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to advanced synthetic routes like the one described in CN101671309B requires a partner with deep technical expertise and proven manufacturing capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel EFCP synthesis are fully realized in practice. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of intermediate meets the exacting standards required for Voriconazole API synthesis. Our commitment to quality assurance means that clients can trust our supply to support their critical drug development and commercialization timelines without compromise.
We invite potential partners to engage with our technical procurement team to discuss how this innovative manufacturing process can be tailored to your specific volume requirements. By requesting a Customized Cost-Saving Analysis, you can quantify the potential economic impact of switching to our optimized EFCP supply chain. We encourage you to contact us today to obtain specific COA data and route feasibility assessments that demonstrate our capability to deliver high-purity intermediates consistently. Let us collaborate to enhance the efficiency and sustainability of your antifungal drug production portfolio.