Scalable Manufacturing of Elagolix Intermediate Using 6-Methyluracil Starting Material
Scalable Manufacturing of Elagolix Intermediate Using 6-Methyluracil Starting Material
The pharmaceutical industry continuously seeks robust and cost-effective synthetic routes for high-value active pharmaceutical ingredients (APIs) and their precursors. A significant advancement in this domain is detailed in patent CN110204498B, which discloses a highly efficient method for synthesizing an Elagolix intermediate. Elagolix, a potent orally active non-peptide gonadotropin-releasing hormone (GnRH) receptor antagonist, represents a critical therapeutic agent for treating endometriosis. The disclosed technology pivots away from traditional, hazardous starting materials, instead utilizing 6-methyluracil as the foundational building block. This strategic shift simplifies the synthetic pathway into five distinct, manageable steps: amino alkylation, halogenation, coupling, benzyl halide substitution, and deprotection. For R&D directors and process chemists, this approach offers a compelling alternative to legacy methods, promising enhanced safety profiles and streamlined purification protocols that are essential for modern GMP manufacturing environments.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Elagolix intermediates has relied on complex and often hazardous chemical transformations. As illustrated in prior art such as Patent CN1819829A, the conventional route typically initiates with 2-fluoro-6-(trifluoromethyl) benzonitrile. This pathway necessitates a borane reduction step, which introduces significant safety risks due to the pyrophoric nature of borane reagents and requires stringent handling protocols. Furthermore, subsequent condensation with urea and cyclization with diketene adds multiple unit operations, increasing the potential for impurity formation and yield loss. Other methods, such as those described in US8765948B2, employ 2-(2-fluoro-3-methoxyphenyl)-ethyl glyoxylate, requiring reduction by sodium borohydride and substitution reactions catalyzed by zinc powder. The use of stoichiometric zinc generates substantial heavy metal waste, complicating downstream processing and environmental compliance. These legacy routes are characterized by long reaction sequences, expensive reagents, and difficult waste management, creating bottlenecks for reliable pharmaceutical intermediate supplier operations aiming for cost efficiency.
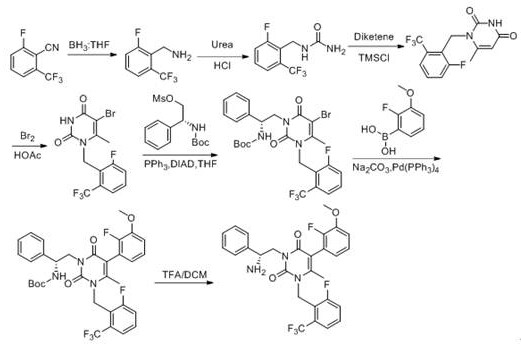
The Novel Approach
In stark contrast, the methodology outlined in CN110204498B leverages 6-methyluracil, a commercially abundant and stable heterocycle, to construct the core scaffold directly. This novel approach eliminates the need for dangerous borane reductions and heavy metal-mediated substitutions. The process is designed with modularity in mind, allowing for precise control over each transformation. By initiating with a pre-formed uracil ring, the synthesis bypasses the energetically demanding cyclization steps required in older patents. The reaction conditions are notably milder, utilizing common organic solvents like N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) rather than exotic or highly toxic media. This simplification not only reduces the operational complexity but also significantly lowers the barrier for commercial scale-up of complex pharmaceutical intermediates. The result is a cleaner reaction profile with fewer by-products, facilitating easier isolation and higher overall purity of the final intermediate, which is crucial for meeting the rigorous quality standards of global regulatory bodies.
Mechanistic Insights into Pd-Catalyzed Suzuki Coupling and Halogenation
A critical component of this synthetic strategy is the palladium-catalyzed Suzuki coupling reaction, which installs the 2-fluoro-3-methoxyphenyl moiety onto the uracil core. The patent specifies the use of palladium acetate in conjunction with tri-tert-butylphosphonium tetrafluoroborate as a ligand system. This specific catalytic combination is chosen for its ability to facilitate oxidative addition and reductive elimination cycles efficiently, even in the presence of potentially coordinating functional groups on the uracil ring. The optimization of catalyst loading, noted at approximately 5 per mill in the preferred embodiments, strikes a balance between reaction kinetics and cost, ensuring high turnover numbers without excessive metal contamination. The coupling proceeds in acetone with a potassium hydroxide base, providing a homogeneous reaction environment that promotes rapid mass transfer. This mechanistic precision ensures that the carbon-carbon bond formation occurs with high regioselectivity, minimizing the formation of homocoupling by-products that often plague Suzuki reactions in complex heterocyclic systems.
Equally important is the halogenation step, which activates the uracil ring for the subsequent coupling. The patent highlights the use of iodine chloride (ICl) as the preferred halogenating agent over elemental bromine or N-bromosuccinimide. Iodine chloride acts as a source of electrophilic iodine, reacting selectively at the C-5 position of the uracil ring. This selectivity is vital because it prevents over-halogenation or degradation of the sensitive Boc-protected amine side chain. The reaction is conducted in methanol at 50 °C, conditions that are mild enough to preserve the stereochemical integrity of the chiral center introduced during the initial amino alkylation. By controlling the molar ratio of the substrate to the halogenating agent at 1:2, the process ensures complete conversion while suppressing the formation of di-halogenated impurities. This level of control is essential for maintaining the high HPLC purity (>97%) reported in the examples, demonstrating a robust mechanism for impurity control throughout the synthesis.
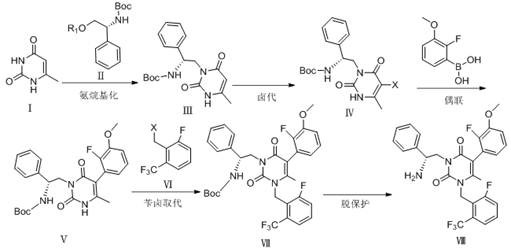
How to Synthesize Elagolix Intermediate Efficiently
The synthesis protocol described in the patent provides a clear roadmap for laboratory and pilot-scale production. The process begins with the amino alkylation of 6-methyluracil, followed by sequential functionalization steps that build molecular complexity with high fidelity. Each step has been optimized for yield and purity, with specific attention paid to workup procedures that avoid chromatographic purification, favoring crystallization and extraction instead. This focus on practical isolation techniques is key to translating the chemistry into a viable manufacturing process. For detailed operational parameters, including exact temperatures, stirring rates, and quenching protocols, operators should refer to the standardized synthesis steps provided below.
- Perform amino alkylation on 6-methyluracil using D-Boc phenylglycinol mesylate and an organic base like 2,6-lutidine in DMF at 55°C.
- Execute halogenation of the resulting compound using iodine chloride in methanol at 50°C to introduce the necessary halogen substituent.
- Conduct a Suzuki coupling reaction with 2-fluoro-3-methoxyphenylboronic acid using a palladium catalyst system in acetone.
- Carry out benzyl halide substitution with 2-fluoro-6-(trifluoromethyl) bromobenzyl using potassium carbonate in tetrahydrofuran.
- Finalize the synthesis through deprotection using trifluoroacetic acid in dichloromethane to yield the final intermediate with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible strategic benefits beyond mere technical elegance. The shift to 6-methyluracil as a starting material fundamentally alters the cost structure of the intermediate. By eliminating the need for specialized, high-cost reagents like borane-THF complexes and stoichiometric zinc powder, the direct material costs are significantly reduced. Furthermore, the simplified workflow reduces the number of unit operations, which translates to lower utility consumption and reduced labor hours per kilogram of product. This efficiency gain allows for cost reduction in API manufacturing, making the final drug product more competitive in the marketplace. The use of common, non-proprietary solvents also mitigates supply risk, ensuring that production is not held hostage by the availability of niche chemicals.
- Cost Reduction in Manufacturing: The elimination of hazardous reducing agents and heavy metal catalysts removes the necessity for expensive waste treatment and metal scavenging processes. Traditional routes often require dedicated downstream processing to remove trace metals to ppm levels, a step that is both costly and time-consuming. By utilizing a palladium catalyst at low loading and avoiding zinc dust, the new method drastically simplifies the purification train. Additionally, the high yields reported in the patent examples (e.g., 96.1% for the coupling step) mean that less raw material is wasted, further driving down the cost of goods sold (COGS). This economic efficiency is critical for maintaining margins in the highly competitive generic and specialty pharmaceutical sectors.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals such as 6-methyluracil, iodine chloride, and standard organic solvents ensures a stable and resilient supply chain. Unlike complex custom synthons that may have single-source suppliers, these raw materials are widely available from multiple global vendors. This diversification reduces the risk of supply disruptions caused by geopolitical issues or manufacturing incidents at a single facility. Moreover, the robustness of the reaction conditions—tolerant to minor variations in temperature and stoichiometry—means that the process can be reliably transferred between different manufacturing sites without significant re-validation, ensuring continuity of supply for downstream API production.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process represents a significant improvement. The avoidance of pyrophoric reagents and the reduction of heavy metal waste align with green chemistry principles, facilitating easier regulatory approval for new manufacturing facilities. The solvents used, such as THF and acetone, are well-understood and can be efficiently recovered and recycled, minimizing the environmental footprint. This compliance advantage is increasingly important as global regulations tighten around pharmaceutical emissions. The process is inherently safer for operators, reducing the likelihood of workplace accidents and associated liabilities, thereby supporting sustainable and scalable commercial operations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These insights are derived directly from the experimental data and comparative examples provided in the patent documentation, offering clarity on process robustness and optimization strategies. Understanding these nuances is vital for technical teams evaluating the feasibility of adopting this route for large-scale production.
Q: Why is 6-methyluracil preferred over 2-fluoro-6-(trifluoromethyl) benzonitrile as a starting material?
A: 6-Methyluracil offers a more direct route to the uracil core structure, eliminating the need for hazardous borane reduction steps and complex cyclization reactions required when starting from benzonitrile derivatives, thereby improving safety and reducing process cost.
Q: What specific catalyst system is optimized for the coupling step in this patent?
A: The patent specifies the use of palladium acetate (0.04g scale in examples) combined with tri-tert-butylphosphonium tetrafluoroborate as a ligand system, which provides high conversion rates and purity compared to other palladium sources like palladium chloride.
Q: How does this method address environmental concerns in pharmaceutical manufacturing?
A: The process utilizes greener solvents such as tetrahydrofuran, acetone, and dichloromethane, and avoids heavy metal waste associated with zinc powder reductions found in prior art, making it more suitable for large-scale commercial production with lower environmental impact.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Elagolix Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthetic routes in the development of life-saving medications. Our team of expert process chemists has extensively analyzed the technology disclosed in CN110204498B and is fully prepared to implement this advanced methodology. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with consistency and precision. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of Elagolix intermediate meets the highest international standards. We are committed to delivering high-purity pharmaceutical intermediates that accelerate your drug development timelines.
We invite you to collaborate with us to leverage this innovative synthesis technology for your projects. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this route can optimize your budget. Please contact us today to request specific COA data and route feasibility assessments. Let us partner with you to bring safer, more affordable treatments to patients worldwide through superior chemical manufacturing excellence.