Advanced Two-Step Synthesis of 5,6,7,8-Tetrahydroimidazo[1,5-a]pyrazine for Pharmaceutical Applications
The pharmaceutical industry constantly seeks more efficient pathways to access complex heterocyclic scaffolds that serve as critical building blocks for drug discovery. A pivotal advancement in this domain is detailed in patent CN101407519A, which discloses a highly practical and economical method for synthesizing 5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine. This specific fused heterocyclic structure is increasingly recognized for its utility in medicinal chemistry, often serving as a rigid bioisostere or a key pharmacophore in various therapeutic agents. The significance of this patent lies not merely in the creation of the molecule itself, but in the fundamental restructuring of the synthetic logic. By moving away from laborious multi-step protection strategies towards a direct alkylation and cyclization approach, the technology addresses the persistent industry pain points of low overall yield and excessive operational complexity. For R&D directors and process chemists, this represents a paradigm shift towards atom-economical design, while supply chain managers will appreciate the reliance on bulk commodity chemicals rather than exotic, expensive reagents.
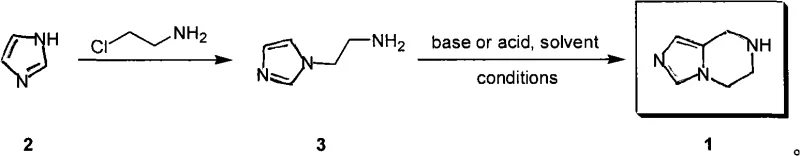
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of 5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine was fraught with inefficiencies that made commercial scale-up economically unviable. As illustrated in the background art of the patent, traditional routes relied heavily on the use of 4-hydroxymethylimidazole as a starting material, which itself requires preparation. More critically, the synthetic pathway necessitated the use of SEM-Cl (2-(trimethylsilyl)ethoxymethyl chloride) for nitrogen protection, followed by oxidation of the hydroxymethyl group to an aldehyde. This was subsequently followed by reductive amination and Boc protection before the final ring closure and deprotection could occur. 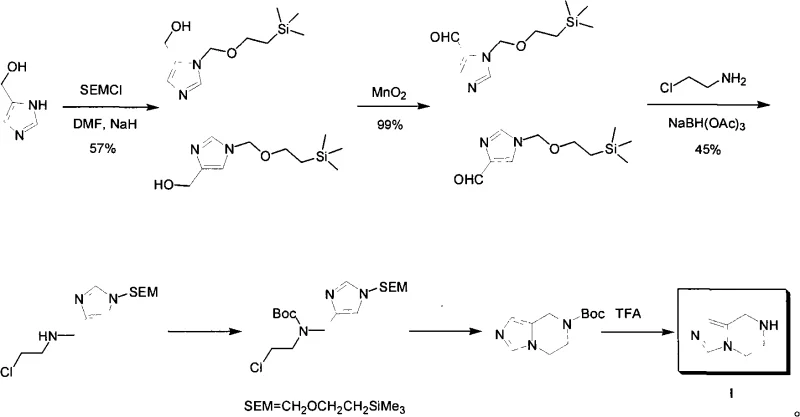 This convoluted sequence results in a significantly elongated production timeline and introduces multiple points of yield loss. Each protection and deprotection step generates stoichiometric waste, increasing the environmental footprint and the cost of goods sold (COGS). Furthermore, reagents like SEM-Cl are moisture-sensitive and relatively expensive, creating supply chain vulnerabilities and driving up the cost of raw materials substantially.
This convoluted sequence results in a significantly elongated production timeline and introduces multiple points of yield loss. Each protection and deprotection step generates stoichiometric waste, increasing the environmental footprint and the cost of goods sold (COGS). Furthermore, reagents like SEM-Cl are moisture-sensitive and relatively expensive, creating supply chain vulnerabilities and driving up the cost of raw materials substantially.
The Novel Approach
In stark contrast, the methodology disclosed in CN101407519A simplifies the entire operation into a concise two-step sequence that bypasses the need for transient protecting groups entirely. The novel approach initiates with the direct N-alkylation of imidazole, a ubiquitous and low-cost industrial feedstock, using chloroethylamine. This reaction efficiently constructs the carbon-nitrogen backbone required for the final structure without the need for oxidation state adjustments. The second step involves a direct cyclization using common formaldehyde equivalents such as paraformaldehyde, methylal, or trioxane. This streamlining eliminates at least four to five discrete unit operations found in the legacy route. By removing the dependency on sensitive silyl protecting groups and avoiding redox manipulations, the new process not only shortens the lead time for batch completion but also drastically improves the robustness of the manufacturing process, making it far more amenable to continuous processing or large-batch reactor campaigns.
Mechanistic Insights into Direct Alkylation and Acid/Base Catalyzed Cyclization
The chemical elegance of this process relies on the inherent nucleophilicity of the imidazole ring and the electrophilicity of the chloroethylamine species. In the first stage, the imidazole nitrogen acts as a nucleophile, attacking the terminal carbon of the chloroethylamine chain. This substitution reaction is facilitated by the presence of a base, such as sodium hydroxide or potassium hydroxide, which serves to deprotonate the imidazole nitrogen, thereby enhancing its nucleophilic character. The use of a phase-transfer catalyst, such as tetrabutylammonium bromide, is often employed to bridge the solubility gap between the inorganic base and the organic substrates in solvents like acetonitrile or tetrahydrofuran (THF). The reaction typically proceeds at elevated temperatures ranging from 80°C to 120°C, ensuring complete conversion to the 2-imidazolylethylamine intermediate. This intermediate is stable and can be isolated as a hydrochloride salt with high purity, as evidenced by the patent's reported yields of up to 95% in optimized embodiments.
The subsequent ring-closing step is a fascinating example of manifold reactivity where the choice of catalyst (acid or base) dictates the mechanism but achieves the same structural outcome. When formaldehyde sources like paraformaldehyde or methylal are introduced, they generate reactive electrophilic species (essentially formaldehyde or iminium ions) in situ. Under basic conditions, the primary amine of the intermediate attacks the formaldehyde to form a hemiaminal or imine, which then undergoes an intramolecular electrophilic aromatic substitution or a Mannich-type cyclization onto the imidazole ring. Alternatively, under acidic conditions (using glacial acetic acid or concentrated HCl), the amine is protonated, but the equilibrium favors the formation of the iminium ion from the formaldehyde source, which is sufficiently electrophilic to be attacked by the electron-rich C2 or C4 position of the imidazole ring (depending on tautomerism), ultimately forging the six-membered pyrazine ring fused to the imidazole. This mechanistic flexibility allows manufacturers to choose conditions that best fit their existing equipment and waste treatment capabilities.
How to Synthesize 5,6,7,8-Tetrahydroimidazo[1,5-a]pyrazine Efficiently
Implementing this synthesis in a pilot or commercial plant requires careful attention to the stoichiometry of the alkylating agent and the selection of the formaldehyde source to maximize yield and minimize byproducts. The patent provides several robust embodiments that serve as a blueprint for process development, highlighting that the reaction is tolerant to various solvents including methanol, ethanol, and water mixtures. The operational simplicity is a key feature; the workup generally involves standard techniques such as filtration to remove inorganic salts, distillation to remove solvents, and precipitation of the final product as a hydrochloride salt using HCl in methanol.
- Perform N-alkylation of imidazole with chloroethylamine hydrochloride in the presence of a base (e.g., NaOH) and a phase-transfer catalyst in solvents like acetonitrile or THF at 80-120°C to obtain 2-imidazolylethylamine.
- React the resulting 2-imidazolylethylamine with a formaldehyde source (such as paraformaldehyde, methylal, or trioxane) in a solvent like methanol or ethanol.
- Facilitate ring closure by adding an acid (e.g., HCl, acetic acid) or base (e.g., NaOH, triethylamine) and heating to 80-120°C, followed by isolation via precipitation or distillation.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers profound strategic benefits that extend beyond simple chemistry. The most immediate impact is seen in the raw material cost structure. By replacing expensive, specialty reagents like SEM-Cl and Boc-anhydride with commodity chemicals like imidazole and chloroethylamine hydrochloride, the direct material cost is significantly reduced. Imidazole is produced on a massive global scale for various industries, ensuring a stable and competitive supply market. This shift mitigates the risk of supply disruptions associated with niche fine chemical suppliers and allows for better long-term price forecasting and budget stability.
- Cost Reduction in Manufacturing: The elimination of protection and deprotection steps translates directly into lower operational expenditures. Fewer steps mean less solvent consumption, reduced energy usage for heating and cooling cycles, and decreased labor hours per kilogram of product. Furthermore, the avoidance of chromatographic purification—relying instead on crystallization and precipitation—drastically lowers the cost of downstream processing. The high yields reported in the patent (up to 85% for the cyclization step) ensure that raw material utilization is maximized, minimizing the cost of wasted inputs and reducing the burden on waste treatment facilities.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to higher reliability in delivery schedules. The process operates at atmospheric pressure and moderate temperatures (80-120°C), which reduces the likelihood of equipment failure or safety incidents that could halt production. Since the starting materials are not hazardous or highly regulated in the same way as some specialized organometallic reagents, logistics and transportation are simplified. This ease of handling ensures that production campaigns can be initiated quickly without lengthy procurement lead times for exotic precursors, thereby reducing the overall lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this green chemistry approach is superior. The atom economy is improved by avoiding the generation of heavy silicate waste from SEM deprotection. The solvents used (methanol, ethanol, acetonitrile) are well-understood and easily recoverable via distillation, supporting a circular solvent management strategy. The ability to produce the target molecule in fewer steps inherently reduces the facility's carbon footprint per unit of output. This aligns with the increasing regulatory pressures on pharmaceutical suppliers to demonstrate sustainable manufacturing practices, making the facility more attractive to top-tier multinational clients who prioritize green supply chains.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its adoption for commercial production. The following questions address common concerns regarding yield optimization, impurity profiles, and scalability, drawing directly from the experimental data provided in the patent documentation.
Q: What are the primary advantages of this synthesis method over traditional routes?
A: The primary advantage is the drastic reduction in synthetic steps. Traditional methods require multiple protection and deprotection steps (SEM, Boc groups) and oxidation/reduction sequences. This novel method achieves the target molecule in just two steps from commodity chemicals, significantly improving total yield and reducing waste.
Q: Is this process suitable for large-scale commercial manufacturing?
A: Yes, the process is highly scalable. It utilizes inexpensive, readily available industrial raw materials like imidazole and chloroethylamine. The reaction conditions (80-120°C, atmospheric pressure) are mild and do not require specialized high-pressure equipment, making it ideal for ton-scale production.
Q: What represents the critical quality control parameter for the intermediate?
A: The purity of the 2-imidazolylethylamine intermediate is crucial, as impurities here can carry through to the final cyclization. The patent demonstrates that simple workups involving filtration and distillation can achieve high purity (e.g., >95% yield in Step 1), ensuring a clean final product.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5,6,7,8-Tetrahydroimidazo[1,5-a]pyrazine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the value of a chemical process lies in its reproducibility and scalability. Our team of expert process chemists has thoroughly analyzed the technology disclosed in CN101407519A and is fully prepared to translate this laboratory-scale success into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from gram-scale experiments to tonnage manufacturing is seamless. Our state-of-the-art facilities are equipped to handle the specific solvent systems and thermal requirements of this alkylation-cyclization sequence, while our rigorous QC labs enforce stringent purity specifications to guarantee that every batch meets the exacting standards required for API synthesis.
We invite potential partners to engage with us to explore how this optimized route can enhance your project's economics. By leveraging our manufacturing capabilities, you can secure a stable supply of this critical intermediate while achieving substantial cost savings. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. We are ready to provide specific COA data from our pilot runs and comprehensive route feasibility assessments to support your regulatory filings and supply chain planning.