Advanced Asymmetric Total Synthesis of (-)-Brazilin for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking efficient pathways to access bioactive natural products with high stereochemical purity. A recent breakthrough detailed in patent CN110776488B presents a robust method for the asymmetric total synthesis of (-)-Brazilin, a homoisoflavonoid derivative renowned for its potent biological activities including anti-tumor, anti-inflammatory, and antioxidant properties. Unlike previous methodologies that struggled with racemization or excessive step counts, this novel approach utilizes a format reagent as an initial raw material to construct chiral carbon atoms connected with hydroxyl groups through precise Mitsunobu Coupling and Sharpless Asymmetric Dihydroxylation reactions. The process culminates in the construction of a complex tetracyclic framework via a key one-step double Friedel-Crafts alkylation, ultimately delivering the target natural product with remarkable efficiency. This technological advancement represents a significant leap forward for manufacturers aiming to secure a reliable supply of high-purity pharmaceutical intermediates.
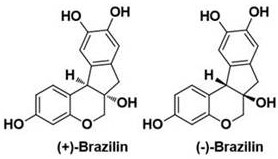
The structural complexity of Brazilin, characterized by its chroman rings and specific phenolic hydroxyl configurations, has historically posed significant challenges for synthetic chemists. As illustrated in the structural comparison, the distinction between enantiomers is critical for biological efficacy, necessitating asymmetric synthesis rather than racemic resolution. The patent explicitly addresses the limitations of prior art, such as the Davis research group's 1993 synthesis of trimethyl etherified derivatives or the Pettus group's 2005 route which required 9 steps for a mere 8.5% total yield of the racemate. Furthermore, while the Qinhong Bo research group achieved a 23% yield, their method still relied on racemization strategies. The current invention overcomes these hurdles by offering a streamlined, enantioselective pathway that minimizes waste and maximizes optical purity, setting a new benchmark for the commercial production of this valuable scaffold.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Brazilin and its analogues has been plagued by inefficient reaction designs that hinder large-scale manufacturing viability. Early approaches often relied on lengthy linear sequences exceeding 9 steps, which inherently compounded yield losses at every stage, resulting in overall yields that were economically unfeasible for industrial applications. Many reported methods produced only racemic mixtures, requiring additional, costly resolution steps to isolate the biologically active (-)-enantiomer, thereby doubling the material throughput requirements without adding value. Additionally, certain legacy protocols utilized expensive or toxic reagents and harsh reaction conditions that complicated safety management and waste disposal, creating substantial barriers for procurement managers focused on cost reduction in pharmaceutical intermediate manufacturing. The low enantiomeric excess (ee) values reported in some literature, such as the 63% ee achieved by Jahng et al., further underscored the inability of conventional methods to consistently deliver the high-purity standards required for drug development.
The Novel Approach
In stark contrast, the methodology disclosed in patent CN110776488B introduces a highly convergent and stereoselective strategy that drastically simplifies the production landscape. By employing a Grignard reagent formation followed by a copper-catalyzed coupling, the route efficiently builds the carbon skeleton early in the sequence. The integration of a Sharpless Asymmetric Dihydroxylation reaction allows for the precise installation of chiral centers with an impressive ee value of 93.6%, eliminating the need for downstream chiral separation. Perhaps most critically, the synthesis constructs the core tetracyclic framework in a single operational step via double Friedel-Crafts alkylation, collapsing what would traditionally be multiple ring-closing events into one high-efficiency transformation. This reduction in linear steps to just eight, combined with a total yield reaching up to 39%, demonstrates a clear path toward scalable and cost-effective production.
Mechanistic Insights into the Asymmetric Synthetic Route
The success of this synthesis lies in the meticulous orchestration of organometallic and oxidative transformations. The process initiates with the generation of a Grignard reagent from 4-bromo veratrole using magnesium and iodine in tetrahydrofuran at 45-55°C, which is then coupled with a halogenated hydrocarbon under the concerted catalysis of lithium chloride and cuprous iodide at 0°C. This step is crucial for establishing the initial carbon-carbon bonds with high fidelity. Subsequent reduction of the ester moiety using diisobutylaluminum hydride (DIBAL-H) at 0°C provides the necessary alcohol intermediate for the Mitsunobu Coupling Reaction. This etherification step, utilizing triphenylphosphine and diisopropyl azodicarboxylate (DIAD), links the aromatic systems effectively. The stereochemical integrity of the molecule is then locked in place during the Sharpless Asymmetric Dihydroxylation, where potassium osmate and the chiral ligand (DHQ)2PHAL facilitate the addition of hydroxyl groups with high enantioselectivity in a tert-butanol and water solvent system.
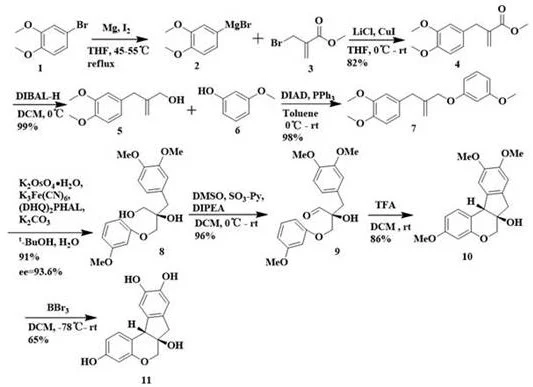
Following the establishment of chirality, the synthesis proceeds through an oxidation step using dimethyl sulfoxide and sulfur trioxide pyridine complex to generate an aldehyde intermediate. This sets the stage for the pivotal double Friedel-Crafts alkylation reaction. By treating the aldehyde precursor with trifluoroacetic acid (TFA) in dichloromethane at room temperature, the molecule undergoes an intramolecular cyclization that simultaneously forms two rings, constructing the rigid tetracyclic core characteristic of Brazilin. The final step involves demethylation using boron tribromide at -78°C, which cleanly removes the methyl protecting groups to reveal the free phenolic hydroxyls of the final product. This mechanistic pathway not only ensures high chemical purity but also minimizes the formation of difficult-to-remove impurities, a key concern for R&D directors overseeing process validation.
How to Synthesize (-)-Brazilin Efficiently
The synthesis of (-)-Brazilin described in this patent offers a reproducible protocol for generating this complex natural product with high optical purity. The route is designed to be operationally simple, utilizing standard laboratory equipment and commonly available solvents such as dichloromethane, tetrahydrofuran, and toluene. For process chemists looking to implement this technology, the detailed experimental section provides specific molar ratios, temperature controls, and workup procedures that ensure consistent results. The following guide outlines the critical phases of the synthesis, from Grignard formation to final purification, serving as a foundational reference for scaling this chemistry from gram to kilogram quantities.
- Preparation of Grignard reagent from 4-bromo veratrole and magnesium, followed by coupling with halogenated hydrocarbon using CuI and LiCl.
- Reduction of ester to alcohol, followed by Mitsunobu coupling and Sharpless Asymmetric Dihydroxylation to establish chiral centers.
- Oxidation to aldehyde, double Friedel-Crafts alkylation to form the tetracyclic framework, and final demethylation with boron tribromide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthetic route offers tangible strategic benefits beyond mere technical elegance. The reliance on conventional chemical reagents and cheap, easily obtainable raw materials significantly lowers the barrier to entry for production, reducing the dependency on exotic or supply-constrained catalysts. The streamlined nature of the 8-step sequence implies a drastic simplification of the manufacturing workflow, which translates to reduced labor hours, lower energy consumption, and minimized solvent usage per kilogram of product. Furthermore, the mild reaction conditions, particularly the room temperature cyclization and the avoidance of cryogenic temperatures except for the final demethylation, enhance process safety and reduce the capital expenditure required for specialized cooling infrastructure.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts in favor of more abundant metals like copper and magnesium, combined with the high overall yield, drives down the cost of goods sold substantially. By avoiding the need for chiral resolution of racemic mixtures, the process effectively halves the material input required to achieve the same amount of active enantiomer, leading to significant raw material savings. Additionally, the high selectivity of the reactions reduces the burden on purification processes, meaning less silica gel and solvent are consumed during chromatography, further optimizing the operational budget.
- Enhanced Supply Chain Reliability: The use of commodity chemicals such as 4-bromo veratrole, magnesium, and standard organic solvents ensures that the supply chain is robust and resistant to disruptions often associated with specialty reagents. The short linear sequence reduces the lead time for production cycles, allowing for faster response to market demand fluctuations. Because the process does not rely on proprietary enzymes or biocatalysts that may have limited shelf-life or availability, manufacturers can maintain consistent inventory levels and guarantee continuity of supply for their downstream clients.
- Scalability and Environmental Compliance: The protocol is explicitly designed with industrial preparation in mind, featuring simple workup procedures like extraction and crystallization that are easily transferable to large-scale reactors. The reduction in step count inherently lowers the E-factor (environmental factor) by generating less waste per unit of product. Moreover, the ability to conduct key bond-forming reactions at ambient pressure and near-ambient temperatures aligns with green chemistry principles, facilitating easier regulatory approval and reducing the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of (-)-Brazilin. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the feasibility and advantages of this specific manufacturing route. Understanding these details is essential for stakeholders evaluating the potential for licensing or contracting the production of this high-value intermediate.
Q: What is the key advantage of this synthesis method for (-)-Brazilin?
A: The method achieves asymmetric total synthesis in only 8 linear steps with a total yield of up to 39% and an ee value of 93.6%, significantly improving upon previous racemic or low-yield routes.
Q: What are the critical reaction conditions for the key cyclization step?
A: The construction of the tetracyclic framework is achieved through a one-step double Friedel-Crafts alkylation reaction using trifluoroacetic acid (TFA) in dichloromethane at room temperature.
Q: Is this process suitable for industrial scale-up?
A: Yes, the patent highlights mild reaction conditions, the use of conventional chemical reagents, and simple operation steps, making it highly suitable for industrial preparation and commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (-)-Brazilin Supplier
The innovative synthetic route for (-)-Brazilin outlined in patent CN110776488B underscores the immense potential for producing this bioactive molecule at a commercial scale with superior quality metrics. At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex synthetic challenges are met with engineering precision. Our commitment to quality is backed by stringent purity specifications and rigorous QC labs that utilize state-of-the-art analytical instrumentation to verify identity, assay, and impurity profiles, guaranteeing that every batch meets the exacting standards required by global pharmaceutical partners.
We invite you to collaborate with us to leverage this advanced technology for your supply chain needs. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this efficient route can optimize your budget. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how we can become your trusted partner for high-purity pharmaceutical intermediates.