Advanced Synthesis of Hydrogenated Pyridine Derivatives for Scalable Antiplatelet API Production
Advanced Synthesis of Hydrogenated Pyridine Derivatives for Scalable Antiplatelet API Production
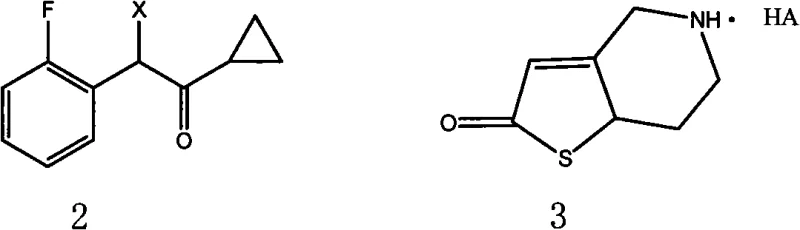
The pharmaceutical industry continuously seeks robust manufacturing pathways for cardiovascular therapeutics, particularly antiplatelet agents that inhibit platelet aggregation. Patent CN101343278A introduces a refined preparation method for hydrogenated pyridine derivatives, specifically 2-acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridine and its salts. This compound class exhibits potent biological activity comparable to established anti-thrombotic medications, offering improved oral absorption and metabolic profiles. The disclosed technology represents a significant leap forward in process chemistry, addressing critical bottlenecks in yield and crystal form control that have historically plagued the production of these complex heterocyclic systems. By optimizing the synthetic route through a convergent strategy, the patent provides a blueprint for generating high-purity intermediates essential for downstream API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art, such as the methodologies documented in EP-192535 and EP-542411, has long been hindered by inefficient synthetic sequences and poor control over solid-state properties. Traditional routes often involve excessive step counts, leading to cumulative yield losses that drastically inflate the cost of goods sold for the final active pharmaceutical ingredient. Furthermore, a persistent challenge in the field has been the inability to consistently isolate the target molecule in a single, stable crystal form. The presence of mixed crystal forms can compromise the bioavailability and shelf-life of the medication, creating significant regulatory hurdles for drug developers. These legacy processes frequently rely on harsh reaction conditions or difficult-to-remove impurities, necessitating complex purification protocols that are ill-suited for cost-effective industrial application.
The Novel Approach
The innovation presented in this patent circumvents these historical deficiencies by employing a modular, convergent synthesis that couples two distinct key intermediates. As illustrated in the reaction scheme below, the process strategically unites an α-cyclopropylcarbonyl-2-fluorobenzyl halide with a 2-oxo-tetrahydrothienopyridine salt. This approach not only streamlines the workflow but also facilitates superior control over the final crystallization process through the use of specific seed crystals. By decoupling the synthesis of the lipophilic side chain from the heterocyclic core, chemists can optimize each fragment independently before the final assembly, ensuring higher overall purity and reproducibility.
Mechanistic Insights into Grignard-Mediated Side Chain Construction
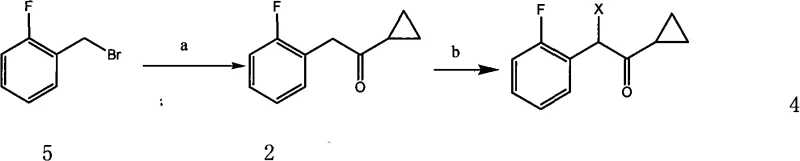
A cornerstone of this improved methodology is the optimized construction of the fluorobenzyl ketone side chain, which serves as the electrophilic partner in the final coupling. The process initiates with the formation of a Grignard reagent from 2-fluorobenzyl bromide and magnesium turnings in anhydrous tetrahydrofuran or diethyl ether. This organometallic species is then reacted with cyclopropyl cyanide to install the cyclopropyl ketone motif, a critical pharmacophore for biological activity. Subsequent halogenation, utilizing reagents such as bromine or N-bromosuccinimide, converts the methylene group adjacent to the carbonyl into a reactive halide. This sequence, depicted in the following diagram, avoids the use of expensive transition metal catalysts and proceeds under relatively mild thermal conditions, typically between 0°C and 60°C, minimizing side reactions and degradation.
Parallel to the side chain synthesis, the heterocyclic core is prepared through a carefully orchestrated sequence of protection, oxidation, and deprotection steps. The tetrahydrothienopyridine scaffold is first protected at the nitrogen atom to prevent unwanted alkylation during subsequent transformations. An oxygen substitution reaction introduces the ketone functionality at the 2-position of the thiophene ring, followed by the removal of the protecting group under acidic conditions to reveal the reactive secondary amine. This multi-step refinement of the core ensures that the nucleophilic nitrogen is available for the final condensation without interference from other functional groups, thereby enhancing the regioselectivity of the coupling reaction and reducing the formation of structural isomers.
How to Synthesize 2-Acetoxy-5-(α-cyclopropylcarbonyl-2-fluorobenzyl) Derivatives Efficiently
The execution of this synthesis requires precise control over reaction parameters to maximize yield and ensure the formation of the desired polymorph. The final assembly involves the condensation of the halogenated side chain with the thienopyridine core in the presence of a base such as triethylamine or pyridine, which acts as both a solvent and an acid scavenger. Following the formation of the tertiary amine linkage, the 2-keto group is esterified using acetic anhydride to yield the final acetate prodrug structure. The detailed operational parameters, including specific molar ratios, solvent choices, and temperature ramps for each stage, are critical for successful replication and are outlined in the standardized guide below.
- Prepare the α-cyclopropylcarbonyl-2-fluorobenzyl halide intermediate via Grignard reaction of 2-fluorobenzyl bromide with magnesium, followed by reaction with cyclopropyl cyanide and subsequent halogenation.
- Synthesize the 2-oxo-2,4,5,6,7,7a-hexahydrothieno[3,2-c]pyridine salt by protecting the amine of the tetrahydrothienopyridine, performing oxygen substitution, and deprotecting under acidic conditions.
- Condense the two key intermediates under alkaline conditions to form the base structure, followed by esterification with acetic anhydride and salt formation to obtain the final crystalline product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement specialists and supply chain managers, the adoption of this patented process offers tangible strategic benefits that extend beyond mere technical feasibility. The streamlined nature of the synthesis directly translates to a reduction in the consumption of raw materials and solvents, effectively lowering the variable costs associated with manufacturing. By eliminating the need for complex transition metal catalysts and reducing the total number of isolation steps, the process minimizes waste generation and simplifies the environmental compliance burden. This efficiency gain allows for a more competitive pricing structure, making the supply of these critical intermediates more resilient against market fluctuations and raw material shortages.
- Cost Reduction in Manufacturing: The elimination of expensive catalytic systems and the reduction in unit operations significantly lower the overall production expenditure. By utilizing readily available starting materials like 2-fluorobenzyl bromide and avoiding proprietary reagents, the process ensures a lean cost structure that supports margin improvement for downstream API producers without compromising on quality standards.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals and standard laboratory equipment enhances the robustness of the supply chain. Since the synthetic route does not depend on scarce or geographically concentrated resources, manufacturers can diversify their supplier base and mitigate the risk of disruptions, ensuring a continuous flow of high-quality intermediates to meet global demand.
- Scalability and Environmental Compliance: The mild reaction conditions and the use of common organic solvents facilitate easy scale-up from pilot plant to commercial production volumes. Furthermore, the simplified workup procedures reduce the volume of hazardous waste requiring treatment, aligning the manufacturing process with increasingly stringent environmental regulations and sustainability goals.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These insights are derived directly from the experimental data and comparative analysis provided within the patent documentation, offering clarity on the practical aspects of adopting this technology for industrial purposes.
Q: How does this patent address the issue of crystal form purity compared to prior art?
A: Unlike previous methods described in EP-542411 which struggled to obtain single crystal forms, this invention utilizes a specific seeding technique during the crystallization of the acid addition salt, ensuring a consistent and pure crystal lattice essential for pharmaceutical stability.
Q: What are the key advantages of the Grignard-based side chain synthesis?
A: The optimized route replaces complex multi-step sequences with a direct Grignard addition to cyclopropyl cyanide followed by halogenation. This significantly reduces the number of unit operations, minimizes waste generation, and improves the overall yield of the critical fluorobenzyl ketone intermediate.
Q: Is this process suitable for large-scale industrial manufacturing?
A: Yes, the reaction conditions are mild, typically ranging from 0°C to 60°C, and utilize common solvents like THF and dichloromethane. The avoidance of extreme temperatures or pressures, combined with robust purification methods like silica gel chromatography and recrystallization, makes it highly adaptable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Hydrogenated Pyridine Derivative Supplier
At NINGBO INNO PHARMCHEM, we leverage deep technical expertise to transform innovative patent methodologies into commercial reality. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to manufacturing plant is seamless and efficient. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of hydrogenated pyridine derivative meets the exacting standards required for pharmaceutical applications, providing our partners with absolute confidence in material quality.
We invite global pharmaceutical companies to collaborate with us to unlock the full potential of this advanced synthesis route. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and comprehensive route feasibility assessments to support your development timelines and help you secure a reliable supply of high-purity intermediates for your antiplatelet drug portfolio.