Advanced Synthesis of 1,3-Azasilane Compounds for Next-Generation Pharmaceutical Intermediates
The pharmaceutical industry is constantly seeking robust, scalable, and cost-effective pathways to access novel heterocyclic scaffolds, particularly those involving bioisosteric replacements like silicon-for-carbon swaps. Patent CN114539304B, published in early 2023, introduces a groundbreaking synthetic methodology for a class of 1,3-azasilane compounds, which serve as critical precursors for constructing sila-indolines. This technology represents a significant leap forward in silicon-nitrogen compound chemistry, addressing long-standing challenges regarding reaction harshness, catalyst toxicity, and substrate availability. By leveraging a sequence of nucleophilic substitutions followed by a unique [3+2] cyclization, this invention provides a reliable route to high-purity pharmaceutical intermediates that were previously difficult to manufacture. For R&D teams and procurement strategists alike, understanding the nuances of this patent is essential for evaluating new supply chain opportunities in the realm of advanced medicinal chemistry.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of silicon-containing nitrogen heterocycles, such as sila-indolines, has been plagued by significant synthetic hurdles. Prior art, including methodologies reported by research groups such as Huang and Driver, relied heavily on the use of precious and toxic transition metal catalysts like Ruthenium (Ru) and Rhodium (Rh). These conventional routes often necessitated the pre-functionalization of substrates with specific silicon-nitrogen groups, a process that is not only synthetically tedious but also limits the scope of applicable starting materials. Furthermore, the reliance on heavy metal catalysis introduces severe complications in downstream processing, particularly regarding the removal of metal residues to meet stringent pharmaceutical purity standards. The environmental footprint of these methods is also considerable, given the toxicity of the catalysts and the often harsh reaction conditions required to drive the intramolecular cyclizations. These factors collectively result in elevated manufacturing costs and extended lead times, creating a bottleneck for the commercial development of silicon-based drug candidates.
The Novel Approach
In stark contrast, the methodology disclosed in CN114539304B offers a streamlined, metal-free alternative that fundamentally reshapes the synthesis landscape for these compounds. The core innovation lies in a two-stage process that begins with the sequential nucleophilic substitution of chlorosilanes with Grignard reagents, followed by coupling with protected sulfonamides. This approach eliminates the need for expensive transition metals entirely, utilizing instead widely available bulk chemicals such as chloromethyldimethylsilyl chloride and benzylmagnesium chloride. The reaction conditions are remarkably mild, typically proceeding in tetrahydrofuran (THF) at temperatures ranging from 0°C to room temperature for the initial substitution, and moderately elevated temperatures (60-90°C) for the subsequent amine coupling. This shift not only drastically simplifies the operational complexity but also enhances the safety profile of the manufacturing process. By avoiding the pre-installation of complex functional groups and utilizing a modular building block strategy, this novel approach ensures strong applicability across a diverse range of substituents, making it an ideal platform for the rapid generation of chemical libraries for drug discovery.
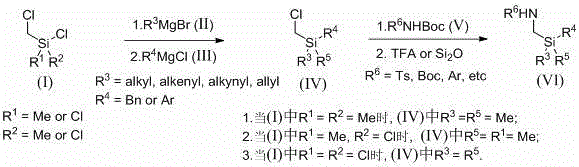
Mechanistic Insights into Nucleophilic Substitution and [3+2] Cyclization
The chemical elegance of this patent lies in its precise control over silicon reactivity to forge carbon-silicon and nitrogen-silicon bonds with high fidelity. The first stage involves the nucleophilic attack of Grignard reagents (R-MgX) on the silicon center of dichlorosilanes. This step is critical for establishing the organic framework around the silicon atom, where parameters such as addition rate (e.g., 10 mL/h) and stoichiometry (1.0:1.0-2.2:1.0-1.2) are tightly controlled to prevent over-reaction or polymerization. Following the formation of the chloromethyl-silyl intermediate, the second stage employs a base-mediated nucleophilic substitution with N-protected amines, such as TsNHBoc. The use of bases like potassium carbonate or cesium carbonate in polar aprotic solvents like DMF facilitates the displacement of the remaining chloride, effectively installing the nitrogen functionality. The final deprotection step using trifluoroacetic acid (TFA) reveals the free amine, yielding the versatile 1,3-azasilane synthon ready for cyclization.
The true mechanistic breakthrough, however, is observed in the subsequent transformation of these 1,3-azasilane compounds into sila-indolines. Unlike previous reports that utilized azetidine precursors for [4+2] ring expansion reactions catalyzed by Palladium, this invention leverages a 1,3-nitrogen-silicon synthon for an intermolecular [3+2] cyclization. In the presence of a benzyne precursor (generated in situ from 2-(trimethylsilyl)phenyl triflate) and a fluoride source or base, the 1,3-azasilane undergoes a unique reaction pathway. It is hypothesized that a hypervalent pentavalent silicon intermediate is formed, which triggers the cleavage of the C(sp3)-Si bond and subsequent ring closure. This mechanism is fundamentally distinct from the C(sp3)-Cl bond cleavage seen in prior art, allowing for the construction of the five-membered benzosilanitrogen heterocycle with exceptional efficiency. This mechanistic divergence not only expands the chemical space accessible to chemists but also provides a cleaner reaction profile with fewer byproducts, directly impacting the purity and yield of the final active pharmaceutical ingredients.
![Mechanism comparison showing [3+2] cyclization versus prior art [4+2] expansion](/insights/img/1-3-azasilane-synthesis-pharma-supplier-20260307194448-042.png)
How to Synthesize 1,3-Azasilane Compounds Efficiently
Implementing this synthesis protocol requires careful attention to reaction parameters to maximize yield and minimize impurities. The patent details specific embodiments, such as Example 5, which outlines the preparation of Compound C (a key 1,3-azasilane intermediate) with a cumulative four-step yield of roughly 10-30% depending on the specific derivatives. The process begins with the Grignard addition in anhydrous THF under inert atmosphere, followed by quenching and extraction. The subsequent coupling with TsNHBoc is performed at 90°C for 5 hours, ensuring complete conversion before the final acidic deprotection. For the cyclization to sila-indoline (Example 7), the reaction utilizes cesium carbonate and 18-crown-6 in ethylene glycol dimethyl ether at 25°C, demonstrating the mildness of the final ring-closing step. While the detailed experimental procedures provide a robust foundation, scaling this chemistry requires optimization of mixing and heat transfer to maintain the precise stoichiometric balances described in the patent text.
- Perform nucleophilic substitution of chloromethylsilanes with Grignard reagents in THF at 0°C to room temperature to form the silyl backbone.
- React the intermediate with protected sulfonamides (e.g., TsNHBoc) under basic conditions (K2CO3, DMF) at 60-90°C.
- Remove the Boc protecting group using TFA or silica gel, followed by [3+2] cyclization with benzyne precursors to yield sila-indolines.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthetic route offers profound advantages for supply chain stability and cost management in the production of pharmaceutical intermediates. The most immediate benefit is the drastic reduction in raw material costs associated with the elimination of precious metal catalysts. Traditional methods relying on Ruthenium, Rhodium, or Palladium incur significant expenses not only for the catalysts themselves but also for the specialized ligands and scavengers required to remove metal traces. By shifting to a base-metal-free protocol using magnesium and silicon reagents, manufacturers can achieve substantial cost savings in the bill of materials. Furthermore, the reagents employed, such as chlorosilanes and Grignard reagents, are commodity chemicals available from multiple global suppliers, mitigating the risk of supply chain disruptions that often plague specialty catalyst markets. This diversification of the supply base enhances procurement resilience and allows for more favorable negotiation leverage.
Enhanced supply chain reliability is further bolstered by the operational simplicity and safety of the process. The reaction conditions are notably mild, avoiding the high pressures or extreme temperatures that often necessitate specialized reactor equipment and rigorous safety protocols. The use of common solvents like THF, DMF, and dichloromethane aligns with standard infrastructure in most fine chemical manufacturing facilities, facilitating easier technology transfer and scale-up. Additionally, the absence of heavy metals simplifies the waste treatment process, reducing the environmental compliance burden and associated disposal costs. This 'green chemistry' aspect is increasingly critical for maintaining social license to operate and meeting the sustainability goals of major pharmaceutical clients. The streamlined workup procedures, involving standard extractions and chromatography, also contribute to shorter cycle times, enabling faster turnaround from raw material intake to finished intermediate delivery.
Scalability and environmental compliance are inherently built into this design, making it highly attractive for commercial scale-up of complex pharmaceutical intermediates. The patent explicitly notes that the reagents are commercially available bulk raw materials that can be stored at room temperature, removing logistical hurdles related to cold chain storage or unstable reagents. The high atom economy of the nucleophilic substitution steps, combined with the efficient [3+2] cyclization, ensures that waste generation is minimized relative to the product output. For supply chain heads, this translates to a more predictable production schedule with fewer batch failures due to catalyst deactivation or sensitivity. The ability to produce high-purity sila-indolines without the need for extensive metal scrubbing steps means that the final product quality is more consistent, reducing the risk of costly rejections during quality control testing. Overall, this technology presents a low-risk, high-reward opportunity for expanding capacity in the silicon-nitrogen compound sector.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this 1,3-azasilane synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in patent CN114539304B, aiming to clarify the practical implications for potential partners and licensees. Understanding these details is crucial for assessing the feasibility of integrating this route into existing manufacturing portfolios.
Q: What are the advantages of this 1,3-azasilane synthesis over traditional methods?
A: Unlike prior art requiring expensive transition metal catalysts like Ruthenium or Rhodium, this method utilizes commercially available Grignard reagents and mild conditions, eliminating heavy metal contamination risks and significantly reducing raw material costs.
Q: How does the [3+2] cyclization mechanism differ from previous approaches?
A: This patent utilizes a unique 1,3-nitrogen-silicon synthon that undergoes intermolecular [3+2] cyclization with benzyne intermediates via a pentavalent silicon species, distinct from the [4+2] expansion reactions seen in older azetidine-based methodologies.
Q: Is this process scalable for industrial production of pharmaceutical intermediates?
A: Yes, the process avoids harsh conditions and hazardous reagents, using standard solvents like THF and DMF. The absence of transition metal catalysts simplifies downstream purification, making it highly suitable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1,3-Azasilane Compounds Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of silicon-based bioisosteres in modern drug design, particularly for targets requiring enhanced metabolic stability and membrane permeability. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory discoveries like those in CN114539304B can be seamlessly transitioned to industrial reality. Our state-of-the-art facilities are equipped to handle the specific solvent systems and reaction conditions required for 1,3-azasilane synthesis, while our rigorous QC labs enforce stringent purity specifications to meet the exacting demands of the global pharmaceutical market. We are committed to delivering high-purity 1,3-azasilane compounds and sila-indoline intermediates that empower your R&D teams to push the boundaries of medicinal chemistry.
We invite you to collaborate with us to unlock the full commercial potential of this technology. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this metal-free route can optimize your COGS. Please contact us to request specific COA data for our available sila-indoline derivatives and to discuss route feasibility assessments for your custom projects. Together, we can accelerate the development of next-generation therapeutics through superior chemical manufacturing.