Advanced Manufacturing of 3'-O-Amino-Ribonucleotides for Next-Gen RNA Therapeutics
Advanced Manufacturing of 3'-O-Amino-Ribonucleotides for Next-Gen RNA Therapeutics
The rapid evolution of RNA therapeutics, including mRNA vaccines and siRNA drugs, has placed unprecedented demand on the supply chain for high-purity nucleotide building blocks. A critical bottleneck in this sector has been the efficient synthesis of 3'-O-amino-ribonucleotides, which serve as essential reversible terminators in next-generation sequencing and enzymatic RNA synthesis. Patent CN113423714A introduces a groundbreaking chemical pathway that addresses the longstanding challenge of regioselectivity in ribonucleoside modification. By leveraging a unique 3'-hydroxy-inverted starting material, this technology enables the precise installation of amino blocking groups without compromising the integrity of the sensitive 2'-hydroxyl moiety. This innovation represents a significant leap forward for manufacturers seeking reliable sources of complex nucleic acid intermediates.
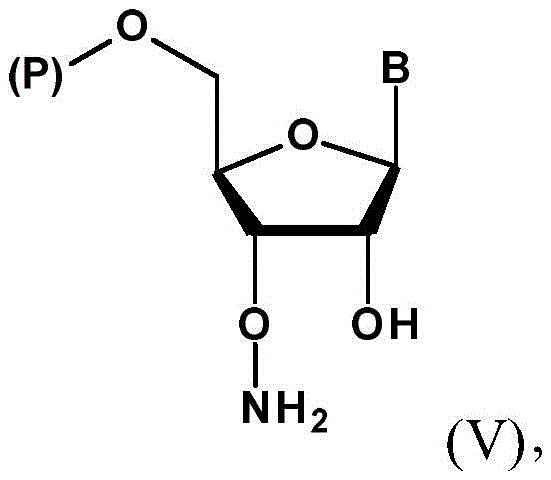
The strategic value of this patent lies in its ability to bypass the limitations of traditional deoxyribonucleoside strategies, which fail when applied to ribose sugars due to steric and electronic interference from the 2'-position. For R&D directors overseeing process development, understanding this mechanistic divergence is crucial for evaluating new suppliers. The disclosed method not only improves overall yield but also simplifies the purification landscape, directly impacting the cost of goods sold (COGS) for downstream applications. As the industry moves towards longer and more complex RNA strands, the availability of such precision-engineered monomers becomes a defining factor in project timelines and commercial viability.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3'-O-amino nucleosides relied heavily on methodologies developed for 2'-deoxyribonucleosides, where the absence of a 2'-hydroxyl group simplifies regioselective functionalization. However, applying these same strategies to ribonucleosides presents severe chemical hurdles. The presence of the vicinal 2'-hydroxyl group creates a competitive environment where protecting group manipulation often leads to migration, elimination, or non-selective acylation. Conventional routes typically involve cumbersome multi-step protection-deprotection sequences, such as Mitsunobu inversion followed by phthalimide insertion, which suffer from low atom economy and generate significant chemical waste. Furthermore, the instability of certain intermediates under standard acidic or basic conditions often necessitates cryogenic temperatures and inert atmospheres, driving up operational costs and limiting batch sizes.
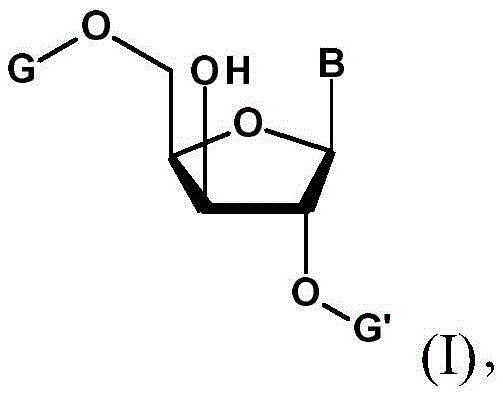
The Novel Approach
The methodology outlined in CN113423714A circumvents these pitfalls by initiating the synthesis with a pre-inverted 3'-hydroxy ribonucleoside scaffold. This strategic starting point allows for a direct sulfonylation at the 3'-position, activating the carbon center for subsequent nucleophilic attack. Unlike traditional methods that struggle to differentiate between the 2' and 3' hydroxyls, this process utilizes specific protecting groups (G and G') at the 2' and 5' positions to sterically shield these sites. The result is a highly selective transformation where the sulfonyl group is installed exclusively at the 3'-oxygen. This streamlined approach reduces the total number of synthetic steps and eliminates the need for harsh inversion reagents late in the sequence, thereby preserving the stereochemical integrity of the sugar ring and improving the overall purity profile of the final API intermediate.
Mechanistic Insights into Sulfonylation-Mediated Amination
The core of this technological advancement resides in the two-step activation and substitution sequence. In the first stage, the 3'-hydroxyl group of the inverted ribonucleoside reacts with a sulfonyl source, such as p-toluenesulfonyl chloride or trifluoromethanesulfonic anhydride, in the presence of a base like triethylamine. This reaction converts the poor leaving group (hydroxyl) into an excellent leaving group (sulfonate ester), creating the reactive intermediate of Formula II. The choice of sulfonyl group is critical; triflyl groups offer higher reactivity for sterically hindered substrates, while tosyl groups provide better stability for isolation. This activation step is conducted under controlled thermal conditions, typically ranging from -10°C to 40°C, to prevent degradation of the nitrogenous base or migration of the silyl protecting groups.
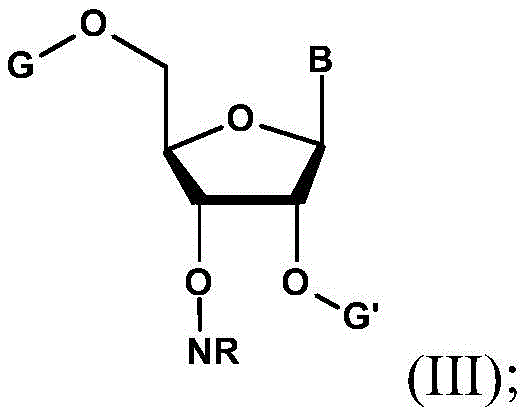
Following activation, the process proceeds to the key amination step where the sulfonate ester undergoes nucleophilic substitution with a hydroxyamino derivative, specifically N-hydroxy-phthalimide or an oxime. This reaction effectively inverts the configuration back to the natural ribose orientation while installing the masked amino group. The use of phthalimido or imino masks is particularly advantageous because they are stable to the conditions used in solid-phase synthesis yet can be cleanly removed later to reveal the free amine. The mechanism likely proceeds via an SN2-type displacement, driven by the strong nucleophilicity of the deprotonated hydroxylamine species. This precise chemical logic ensures that the resulting 3'-O-amino-ribonucleotide precursor (Formula III) possesses the exact stereochemistry required for enzymatic recognition by polymerases, a critical quality attribute for therapeutic efficacy.
How to Synthesize 3'-O-Amino-Ribonucleotides Efficiently
Implementing this synthesis route requires careful control of reaction parameters to maximize yield and minimize impurities. The process begins with the preparation of the inverted starting material, followed by the sequential sulfonylation and amination steps described in the mechanistic section. Operators must pay close attention to the stoichiometry of the sulfonylating agent and the base, as excess reagents can lead to over-sulfonylation or base degradation. The subsequent conversion to the final triphosphate involves a series of deprotection and phosphorylation events that must be orchestrated to avoid hydrolysis of the sensitive phosphate esters. For a detailed breakdown of the specific reagents, solvent systems, and workup procedures required to execute this chemistry at scale, please refer to the standardized protocol below.
- React 3'-hydroxy-inverted ribonucleosides with a sulfonyl source to activate the 3'-position.
- Perform nucleophilic substitution using N-hydroxy-phthalimide or oxime derivatives to install the masked amino group.
- Execute deprotection and phosphorylation sequences to yield the final 3'-O-amino-ribonucleotide triphosphate.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented process translates into tangible operational improvements beyond mere chemical elegance. The primary advantage lies in the significant reduction of process complexity. By eliminating the need for multiple protection and deprotection cycles associated with traditional Mitsunobu-based routes, the manufacturing timeline is drastically compressed. This efficiency gain directly correlates to lower labor costs and reduced consumption of expensive reagents and solvents. Furthermore, the ability to perform key steps in a 'one-pot' fashion minimizes material loss during isolation and purification, leading to higher overall mass throughput. This robustness makes the supply of these critical intermediates more predictable and less susceptible to the variability that often plagues complex nucleoside synthesis.
- Cost Reduction in Manufacturing: The streamlined nature of this synthetic route offers substantial cost savings by removing the requirement for transition metal catalysts or exotic reagents that demand expensive removal protocols. The reliance on commodity chemicals like sulfonyl chlorides and hydrazine ensures that raw material costs remain stable and low. Additionally, the improved selectivity reduces the burden on downstream purification processes, such as chromatography, which are often the most cost-intensive part of nucleotide manufacturing. By simplifying the impurity profile, manufacturers can achieve higher yields of saleable product per batch, effectively lowering the unit cost of the final high-purity pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: Supply continuity is paramount for RNA drug developers, and this process enhances reliability by utilizing widely available starting materials and reagents. Unlike routes dependent on scarce chiral pool resources or custom-synthesized catalysts, the inputs for this method are commercially accessible from multiple global vendors. This diversification of the supply base mitigates the risk of shortages and price volatility. Moreover, the robustness of the chemistry allows for flexible manufacturing schedules, enabling suppliers to respond more rapidly to fluctuations in market demand without compromising on the stringent quality standards required for clinical-grade materials.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, this methodology aligns well with green chemistry principles. The reduction in step count inherently lowers the E-factor (mass of waste per mass of product), reducing the volume of hazardous waste requiring disposal. The solvents employed, such as THF, methanol, and dichloromethane, are well-understood in industrial settings and can be efficiently recovered and recycled. This environmental efficiency not only lowers disposal costs but also simplifies regulatory compliance regarding solvent residues and emissions. Consequently, scaling this process from kilogram to metric ton quantities presents fewer engineering challenges compared to more fragile synthetic pathways.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear picture of the method's capabilities and limitations. Understanding these nuances is essential for technical teams evaluating the feasibility of integrating these intermediates into their own nucleic acid assembly pipelines.
Q: How does this process overcome the selectivity issues of 2'-hydroxyl groups?
A: The patented method utilizes a specific 3'-hydroxy-inverted ribonucleoside starting material where the stereochemistry is pre-adjusted. By employing bulky protecting groups like TBS or TOM at the 2' and 5' positions, the process sterically hinders unwanted reactions at the 2'-site, ensuring the sulfonylation and subsequent amination occur exclusively at the 3'-position.
Q: Can this synthesis be scaled for commercial production of RNA intermediates?
A: Yes, the process is designed for scalability. Key steps such as sulfonylation and amination can be performed in a 'one-pot' sequence without isolating unstable intermediates. The use of robust reagents like triethylamine and common organic solvents facilitates large-scale manufacturing suitable for industrial nucleic acid synthesis.
Q: What are the advantages of using phthalimido or imino masking groups?
A: These masking groups provide exceptional stability during the harsh conditions of oligonucleotide chain elongation. Unlike traditional blocking groups, the amino functionality remains inert until specifically cleaved, preventing premature chain termination or side reactions, which is critical for maintaining high fidelity in enzymatic or chemical RNA synthesis.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3'-O-Amino-Ribonucleotides Supplier
As the global demand for RNA-based therapeutics continues to surge, securing a dependable supply of high-quality nucleotide building blocks is more critical than ever. NINGBO INNO PHARMCHEM stands at the forefront of this industry, leveraging advanced synthetic methodologies like the one described in CN113423714A to deliver superior products. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous volume requirements of large-scale clinical and commercial programs. We maintain stringent purity specifications through our rigorous QC labs, guaranteeing that every batch of 3'-O-amino-ribonucleotides meets the exacting standards necessary for sensitive enzymatic applications.
We invite pharmaceutical and biotech partners to collaborate with us to optimize their supply chains. By leveraging our technical expertise, we can provide a Customized Cost-Saving Analysis tailored to your specific project needs, identifying opportunities to reduce COGS without sacrificing quality. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you accelerate your RNA therapeutic development with reliable, scalable, and cost-effective chemical solutions.