Revolutionizing Sofosbuvir Intermediate Production with a Novel Three-Step Uridine Route
Revolutionizing Sofosbuvir Intermediate Production with a Novel Three-Step Uridine Route
The pharmaceutical industry's relentless pursuit of efficient Hepatitis C treatments has placed immense pressure on the supply chain for key antiviral intermediates. Patent CN111018844B introduces a groundbreaking preparation method for a sofosbuvir key intermediate that fundamentally alters the economic and technical landscape of API manufacturing. By shifting the synthetic starting point to uridine, a cheap and abundant natural nucleoside, this invention bypasses the convoluted multi-step sequences that have historically plagued production. The disclosed methodology not only streamlines the process into a concise three-step sequence but also addresses critical pain points regarding chiral purity and environmental safety. For R&D directors and procurement strategists alike, this represents a pivotal opportunity to optimize the cost structure of nucleotide analog production while ensuring a robust, scalable supply of high-purity materials essential for next-generation antiviral therapies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of sofosbuvir intermediates has been fraught with inefficiencies, primarily characterized by excessive step counts and the reliance on hazardous reagents. As illustrated in the prior art routes, traditional methods often commence with cytidine or complex dioxolane derivatives, necessitating a tedious cascade of over ten distinct chemical transformations. These legacy pathways involve repetitive cycles of benzoyl protection, silyl ether protection, oxidation, and subsequent deprotection, leading to poor atom economy and significant material loss at each stage. Furthermore, the introduction of the critical fluorine atom in older methodologies frequently requires dangerous reagents such as methyl iodide and phosphorus tribromide, which pose severe neurotoxic and respiratory risks to personnel. The cumulative effect of these factors is a manufacturing process that is not only prohibitively expensive due to low overall yields but also environmentally burdensome, creating substantial challenges for waste management and regulatory compliance in modern GMP facilities.
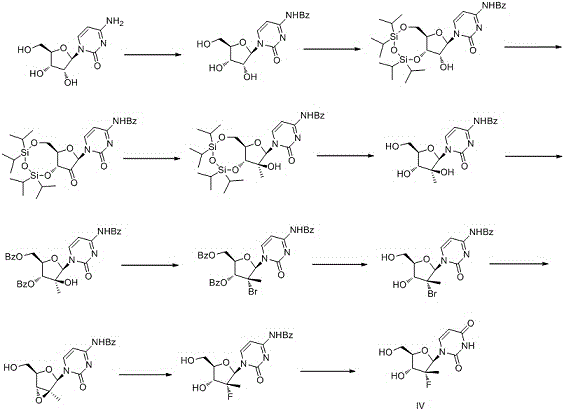
The Novel Approach
In stark contrast to the cumbersome legacy pathways, the novel approach detailed in the patent leverages the intrinsic stereochemical wealth of uridine to achieve a dramatic simplification of the synthetic architecture. This innovative strategy condenses the entire production sequence into merely three high-yielding steps, effectively eliminating the need for repetitive protecting group manipulations that drive up costs in traditional synthesis. By utilizing 1,3-dichloro-1,1,3,3-tetraisopropyl disiloxane for selective silyl protection, the process efficiently masks the 5'-OH and 3'-OH positions, setting the stage for a highly convergent downstream transformation. The true brilliance of this route lies in its ability to construct the complex fluoro-olefin motif through a direct, one-pot fluorination and deprotection sequence, thereby avoiding the chiral erosion associated with multiple inversion steps. This streamlined topology not only accelerates the time-to-market for the intermediate but also establishes a foundation for a reliable sofosbuvir intermediate supplier to deliver consistent quality at a fraction of the historical cost.
Mechanistic Insights into One-Pot Oxidation and Stereoselective Fluorination
The core technical advancement of this patent resides in the sophisticated orchestration of tandem reactions that maximize atomic utilization while maintaining rigorous stereocontrol. The second step involves a seamless one-pot operation where the 2'-hydroxyl group of the silyl-protected uridine is oxidized to a carbonyl species using Jones reagent or similar transition metal oxidants, followed immediately by a Wittig olefination without isolation. This telescoping of oxidation and carbon-carbon bond formation minimizes handling losses and solvent consumption, utilizing methyl triphenyl phosphonium bromide to install the exocyclic methylene group with high fidelity. The reaction conditions are meticulously optimized, employing solvents like dichloromethane or DMSO and bases such as pyridine or NaH to ensure complete conversion within a narrow temperature window of 20-50°C. This mechanistic efficiency is crucial for maintaining the integrity of the sensitive nucleobase and sugar moieties, preventing degradation pathways that often plague nucleoside chemistry.
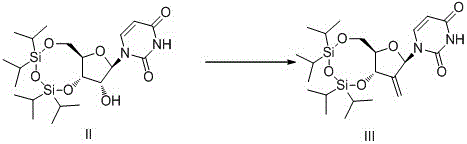
Furthermore, the final fluorination step represents a masterclass in reagent engineering and stereoselectivity. The patent discloses the use of a stabilized hydrofluoric acid complex, specifically formed with potassium bisulfate, which acts as a hydrogen bond acceptor to modulate the acidity and reactivity of the fluoride source. This unique reagent system facilitates the Markovnikov addition of fluorine across the double bond generated in the previous step, adhering strictly to space induction principles to secure the desired stereochemistry at the anomeric center. Crucially, this fluoro-system exhibits remarkable tolerance towards hydroxyl groups, enabling a concurrent deprotection of the silyl ethers in the same reaction vessel. This one-pot fluorination and deprotection capability eliminates the need for separate acidic or basic cleavage steps, thereby reducing the potential for epimerization and ensuring that the final product retains the high chiral purity inherited from the uridine starting material, a critical parameter for the biological efficacy of the resulting antiviral drug.
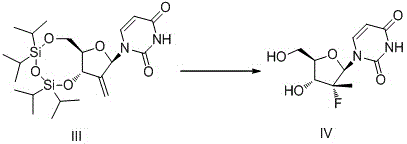
How to Synthesize Sofosbuvir Key Intermediate Efficiently
The implementation of this novel synthetic route requires precise adherence to the reaction parameters defined in the patent to ensure optimal yield and purity profiles. The process begins with the careful selection of base and stoichiometry for the initial silylation, followed by the strictly controlled addition of oxidants and phosphonium salts in the second stage. The final fluorination demands low-temperature control and the specific preparation of the HF-KHSO4 complex to manage the exothermic nature of the addition. While the general chemistry is robust, scaling these reactions requires attention to heat transfer and mixing efficiency, particularly during the exothermic oxidation and fluorination phases. The detailed standardized synthesis steps below outline the specific operational protocols derived from the patent examples, providing a clear roadmap for process chemists to replicate this high-efficiency pathway in a pilot or production setting.
- Protect uridine with 1,3-dichloro-1,1,3,3-tetraisopropyl disiloxane under alkaline conditions to form the bis-silyl ether.
- Perform a one-pot oxidation of the 2'-hydroxyl followed immediately by a Wittig reaction with methyl triphenyl phosphonium bromide.
- Execute a one-pot fluorination using a stabilized hydrofluoric acid complex followed by deprotection to yield the final fluoro-olefin intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this uridine-based synthesis offers transformative benefits for procurement managers and supply chain heads tasked with securing cost-effective and reliable raw materials. The drastic reduction in synthetic steps from over ten to just three inherently translates to a significant reduction in manufacturing costs, driven by lower labor requirements, reduced solvent consumption, and minimized utility usage. By eliminating the need for expensive and toxic reagents like methyl iodide and phosphorus tribromide, the process not only lowers the direct material cost but also mitigates the substantial overhead associated with hazardous waste disposal and worker safety monitoring. This economic efficiency allows for a more competitive pricing structure for the final API, enabling pharmaceutical companies to expand market access for Hepatitis C treatments while maintaining healthy margins in a price-sensitive generic landscape.
- Cost Reduction in Manufacturing: The streamlined three-step protocol eliminates the cumulative yield losses typical of long linear syntheses, resulting in a substantially higher overall throughput of the key intermediate. The use of uridine, a commodity fermentation product, as the starting material ensures a stable and low-cost feedstock supply, insulating the production process from the volatility associated with specialized chiral building blocks. Furthermore, the one-pot nature of the oxidation/Wittig and fluorination/deprotection sequences removes the need for intermediate isolation and purification, which are often the most resource-intensive operations in fine chemical manufacturing, leading to drastic operational expenditure savings.
- Enhanced Supply Chain Reliability: Relying on widely available reagents such as tetraisopropyl disiloxane and methyl triphenyl phosphonium bromide significantly de-risks the supply chain compared to routes dependent on bespoke or regulated precursors. The mild reaction conditions, operating between 0°C and 50°C, reduce the dependency on specialized cryogenic equipment or high-pressure reactors, allowing for flexible production scheduling across a broader range of manufacturing sites. This operational flexibility ensures consistent delivery timelines and reduces the lead time for high-purity pharmaceutical intermediates, safeguarding the continuity of downstream API production against logistical disruptions.
- Scalability and Environmental Compliance: The avoidance of heavy metal catalysts and highly toxic halogenating agents aligns perfectly with modern green chemistry principles and stringent environmental regulations. The process generates a cleaner impurity profile, simplifying the downstream purification burden and reducing the volume of organic waste streams that require treatment. This environmental compatibility facilitates easier regulatory approval for the manufacturing site and supports the long-term sustainability goals of global pharmaceutical partners, making the commercial scale-up of complex nucleoside analogs both technically feasible and socially responsible.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel uridine-based pathway. These insights are derived directly from the experimental data and comparative analysis presented in the patent documentation, aiming to clarify the practical implications for industrial adoption. Understanding these nuances is essential for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the long-term value proposition of this manufacturing route.
Q: How does the new uridine-based route improve chiral purity compared to traditional methods?
A: The new route leverages the inherent chirality of uridine, preserving three of the four required chiral centers from the starting material. Unlike prior art which requires multiple chiral inversions to introduce fluorine, this method constructs the fluorinated chiral center in a single highly stereoselective step, drastically reducing chiral impurities.
Q: What are the safety advantages of the fluorination reagent used in this patent?
A: The process utilizes a potassium bisulfate-hydrofluoric acid complex. This stabilization method increases the effective acidity of the hydrofluoric acid while making it easier to handle compared to free HF gas or highly toxic reagents like methyl iodide and phosphorus tribromide used in older routes, significantly improving operator safety.
Q: Is this synthesis suitable for large-scale industrial production?
A: Yes, the process is explicitly designed for scalability. It reduces the synthetic sequence from over ten steps to just three, utilizes cheap and readily available raw materials like uridine, and employs mild reaction conditions (0-50°C) that are easily manageable in standard stainless steel reactors without requiring cryogenic temperatures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sofosbuvir Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of efficient intermediate synthesis in the global fight against viral diseases. Our team of expert process chemists has thoroughly analyzed the technological breakthroughs in patent CN111018844B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this innovative uridine route to life. We are committed to delivering high-purity sofosbuvir intermediates that meet the most stringent purity specifications, leveraging our rigorous QC labs to ensure every batch conforms to the highest international standards. Our state-of-the-art facilities are equipped to handle the specific reagent requirements and one-pot processing conditions described in the patent, ensuring a seamless transition from laboratory discovery to full-scale industrial supply.
We invite forward-thinking pharmaceutical partners to collaborate with us to unlock the full potential of this cost-effective synthesis. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our advanced manufacturing capabilities can drive down your total cost of ownership while securing a resilient supply chain for your critical antiviral programs.