Scalable Stereoselective Synthesis of Chiral Pyrimidine Intermediates for Pharmaceutical Applications
The pharmaceutical industry continuously demands more efficient and scalable routes for complex chiral intermediates, particularly for potent chemokine receptor antagonists. Patent CN112218853A discloses a groundbreaking stereoselective process for the preparation of chiral 2-[(hetero)aralkylthio]pyrimidines, addressing critical bottlenecks in purity and scalability. This technology enables the production of key intermediates, such as 6-amino-2-{[(1S)-1-phenylethyl]thio}pyrimidin-4-ol, with exceptional chiral purity exceeding 99% without relying on preparative chiral HPLC. The strategic shift from traditional halogenated alkylating agents to carefully engineered sulfonate esters represents a significant leap forward in process chemistry. By controlling the reaction environment to minimize free anions that cause racemization, this method ensures robust stereochemical integrity. 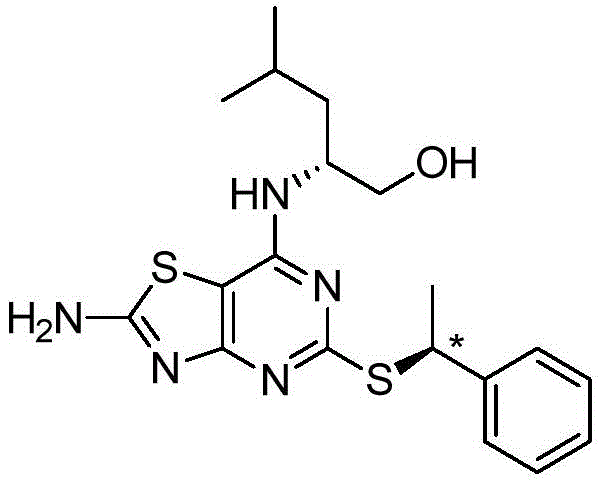 For procurement and supply chain leaders, this translates to a reliable pharmaceutical intermediate supplier capability that reduces dependency on costly purification technologies while maintaining stringent quality standards.
For procurement and supply chain leaders, this translates to a reliable pharmaceutical intermediate supplier capability that reduces dependency on costly purification technologies while maintaining stringent quality standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral benzyl sulfide pyrimidines has relied on the reaction of pyrimidine thiols with chiral chloro-alkanes, such as (R)-1-chloroethylbenzene. While feasible on a laboratory scale, this approach presents severe challenges for commercial manufacturing. The primary issue is the difficulty in controlling stereochemistry; the reaction often yields a mixture of diastereomers, typically around a 9:1 ratio favoring the desired isomer. To achieve the requisite purity for active pharmaceutical ingredients (APIs), manufacturers are forced to employ preparative chiral HPLC. This technique is notoriously expensive, difficult to scale, and results in significant yield loss due to the separation process. Furthermore, the use of chloro-alkanes can lead to racemization via competing mechanisms or multiple inversions if free chloride ions accumulate in the reaction mixture. 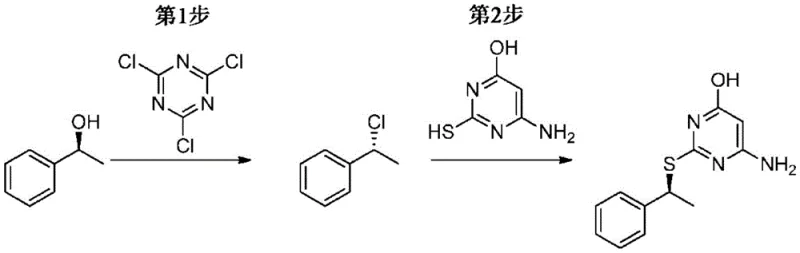 Consequently, the overall yield of conventional processes is often compromised, and the cost of goods sold (COGS) remains prohibitively high for large-scale production.
Consequently, the overall yield of conventional processes is often compromised, and the cost of goods sold (COGS) remains prohibitively high for large-scale production.
The Novel Approach
The patented process introduces a sophisticated alternative by utilizing chiral sulfonate esters, specifically mesylates or tosylates, as the alkylating agents instead of chlorides. This method involves reacting a chiral alcohol with a sulfonating agent in a specific solvent system where the by-product salt is insoluble. For instance, using methanesulfonyl chloride and triethylamine in methyl tert-butyl ether (MTBE) precipitates triethylamine hydrochloride, which is then filtered off. This removal of the leaving group anion (chloride) from the solution is critical; it prevents the anion from participating in undesirable nucleophilic substitutions that would erode chiral purity. The subsequent reaction with the pyrimidine thiolate anion proceeds via a clean bimolecular nucleophilic substitution (SN2) mechanism. This ensures a predictable inversion of configuration and delivers the product with high enantiomeric excess directly from the reactor. 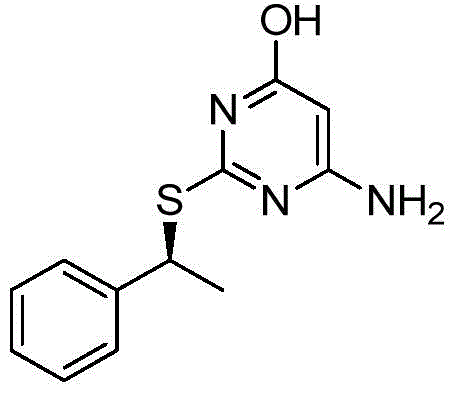 This approach eliminates the need for chiral chromatography, offering a streamlined pathway suitable for multi-kilogram to ton-scale manufacturing.
This approach eliminates the need for chiral chromatography, offering a streamlined pathway suitable for multi-kilogram to ton-scale manufacturing.
Mechanistic Insights into Sulfonate-Mediated Stereoselective Substitution
The core innovation lies in the meticulous control of the reaction medium during the formation of the sulfonate ester (Formula II). The process dictates that the sulfonation step (i) occurs in a solvent (S1) where the salt formed between the base (B1) and the leaving group of the sulfonating agent is insoluble. When methanesulfonyl chloride is used with triethylamine, the resulting triethylamine hydrochloride precipitates in MTBE. By filtering off this solid before the alkylation step, the concentration of free chloride ions in the solution is drastically reduced. This is mechanistically vital because free chloride ions act as nucleophiles that can attack the chiral center, potentially reversing the stereochemistry multiple times (racemization) before the desired thiolate attacks. By removing the chloride source, the process locks in the stereochemistry of the starting alcohol.
Following the isolation or in-situ generation of the sulfonate, the second step involves the nucleophilic attack by the pyrimidine thiolate anion (Formula IV). This species is generated by deprotonating the mercaptopyrimidine (Formula V) with a strong base like sodium hydroxide in a polar aprotic solvent such as DMF. To ensure high purity, a reducing agent like sodium borohydride (NaBH4) is often added to reduce any disulfide dimers (Formula VI) back to the reactive thiolate. The combination of the sulfonate solution with the thiolate solution triggers an SN2 reaction. Since SN2 reactions proceed with Walden inversion, the absolute configuration of the final product is opposite to that of the starting sulfonate. This predictability allows manufacturers to select the appropriate enantiomer of the starting alcohol to obtain the desired product stereochemistry with high fidelity, consistently achieving chiral purities greater than 99%.
How to Synthesize 6-Amino-2-{[(1S)-1-phenylethyl]thio}pyrimidin-4-ol Efficiently
The synthesis of this critical intermediate is achieved through a convergent two-step sequence that maximizes yield and optical purity. The process begins with the activation of (R)-1-phenylethanol via mesylation in MTBE at low temperatures, followed by filtration to remove the amine salt. In parallel, the pyrimidine core is activated as a thiolate anion in DMF. These two streams are then combined under controlled conditions to effect the coupling. The crude product is subsequently purified through a simple precipitation or crystallization step, avoiding complex chromatographic separations. This operational simplicity makes the route highly attractive for industrial adoption.
- React a chiral alcohol with a sulfonating agent (e.g., methanesulfonyl chloride) in a solvent like MTBE with a base to form an insoluble salt by-product, ensuring high stereochemical integrity.
- Prepare a pyrimidine thiolate anion by treating a mercaptopyrimidine with a base (e.g., NaOH) in a polar aprotic solvent like DMF, optionally reducing disulfides with NaBH4.
- Combine the sulfonate solution with the thiolate solution to effect an SN2 substitution, resulting in inversion of configuration and high chiral purity product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this stereoselective sulfonate route offers substantial strategic benefits over legacy chloro-alkylation methods. The most significant advantage is the elimination of preparative chiral HPLC, which is a major cost driver and a bottleneck for supply continuity. By achieving high chiral purity through crystallization and reaction control alone, manufacturers can significantly reduce processing time and solvent consumption. This simplification of the downstream processing directly contributes to cost reduction in pharmaceutical intermediate manufacturing. Furthermore, the reagents used, such as methanesulfonyl chloride and common organic bases, are commodity chemicals with stable supply chains, reducing the risk of raw material shortages.
- Cost Reduction in Manufacturing: The removal of chiral HPLC purification steps drastically lowers the cost of goods. Chromatographic separations require expensive stationary phases, large volumes of high-grade solvents, and specialized equipment, all of which add significant overhead. By replacing this with a filtration and crystallization workflow, the process becomes inherently more economical. Additionally, the higher overall yield resulting from the avoidance of purification losses means less starting material is required per kilogram of final product, further optimizing material costs.
- Enhanced Supply Chain Reliability: The robustness of the SN2 substitution mechanism ensures consistent batch-to-batch quality. In traditional routes, slight variations in reaction conditions can lead to fluctuating diastereomeric ratios, necessitating variable and unpredictable purification efforts. This new method provides a stable platform for production, allowing for more accurate forecasting and inventory planning. The use of standard solvents like MTBE and DMF, which are widely available globally, also mitigates supply chain risks associated with specialty solvents.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind. The precipitation of the amine salt in the first step is easily handled by standard filtration equipment (e.g., nutsche filters) even at large scales. Moreover, the reduction in solvent usage and the elimination of chromatography waste streams align with green chemistry principles. This facilitates easier regulatory approval and reduces the environmental footprint of the manufacturing site, a key consideration for modern pharmaceutical supply chains.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this stereoselective synthesis technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing a clear understanding of the process capabilities and limitations.
Q: How does this process avoid racemization compared to traditional chloro-alkylation?
A: Traditional methods using chloro-alkanes often suffer from competing SN1 pathways or multiple inversions leading to racemization. This novel process utilizes a sulfonate ester leaving group in a solvent system (MTBE) where the base salt is insoluble, minimizing free anions that cause racemization, and relies on a clean SN2 mechanism.
Q: Is chiral HPLC purification required for the final intermediate?
A: No. A key advantage of this invention is achieving chiral purity greater than 99% directly from the reaction and crystallization steps, eliminating the need for expensive and non-scalable chiral HPLC purification typically required in prior art routes.
Q: What are the critical reaction conditions for the sulfonation step?
A: The sulfonation should be conducted at low temperatures (e.g., -10°C to 20°C) in a solvent like methyl tert-butyl ether (MTBE) using a base like triethylamine. Crucially, the solvent must be chosen such that the resulting amine salt (e.g., triethylamine hydrochloride) is insoluble and can be filtered off to prevent side reactions.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Pyrimidine Intermediates Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity chiral intermediates in the development of next-generation therapeutics. Our technical team has extensively analyzed the process disclosed in CN112218853A and is fully equipped to translate this laboratory-scale innovation into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project moves seamlessly from pilot plant to full-scale manufacturing. Our facilities are supported by rigorous QC labs capable of verifying stringent purity specifications, including chiral HPLC analysis to confirm enantiomeric excess levels exceeding 99%.
We invite you to collaborate with us to leverage this advanced synthetic route for your API projects. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential partners to contact us to request specific COA data and route feasibility assessments, ensuring that your supply chain is built on a foundation of technical excellence and economic efficiency.