Advanced Synthesis of Ticagrelor Impurity VIII for Global Pharmaceutical Quality Control
The pharmaceutical industry continuously demands higher standards for impurity profiling to ensure the safety and efficacy of active pharmaceutical ingredients. Patent CN110759917B introduces a groundbreaking preparation method for Ticagrelor Impurity VIII, a critical reference standard required for monitoring the synthesis of the anticoagulant Ticagrelor. This novel synthetic route addresses the longstanding challenge of obtaining high-purity impurity standards without resorting to complex purification techniques. By utilizing cheap and easily obtained urea as a key raw material combined with a specialized condensing agent composition, the process achieves exceptional conversion rates and yields under mild reaction conditions. The ability to reach 98% purity without column chromatography represents a significant technical advancement, facilitating better quality control in the manufacturing of Ticagrelor bulk drugs. This development is particularly vital for regulatory compliance and ensuring patient safety in cardiovascular treatments.
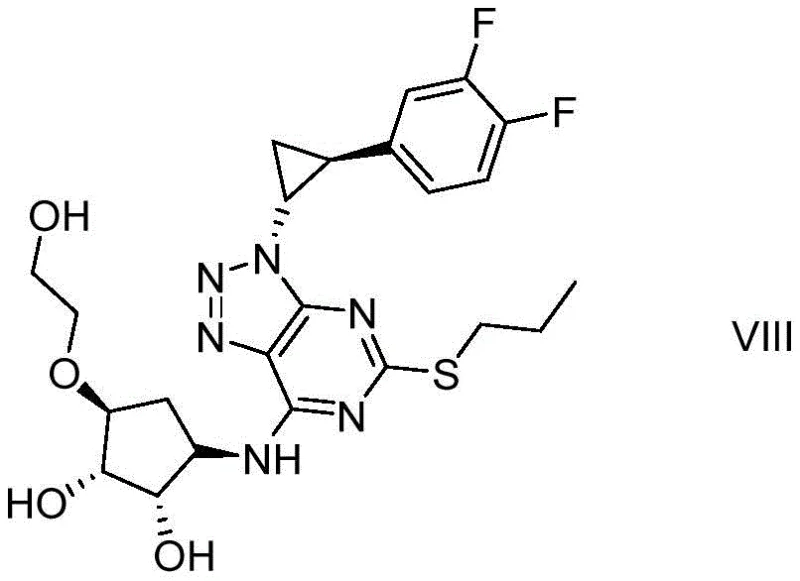
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Ticagrelor involves complex multi-step reactions where substrates such as TG-SM-1, TG-SM-2, and TG-SM-3 may not react completely, leading to the formation of various impurities. These impurities, including Compound VIII, often exist in trace amounts within the final product, posing significant challenges for detection and monitoring. The conventional lack of a dedicated synthesis method for Impurity VIII means that manufacturers struggle to obtain sufficient quantities of high-purity reference substances. This scarcity hampers the ability to accurately monitor impurity levels during the main drug synthesis, potentially compromising the quality control of the Ticagrelor bulk drug. Furthermore, traditional methods often rely on expensive reagents or harsh conditions that result in lower yields and necessitate tedious purification processes like column chromatography, which are not feasible for large-scale production.
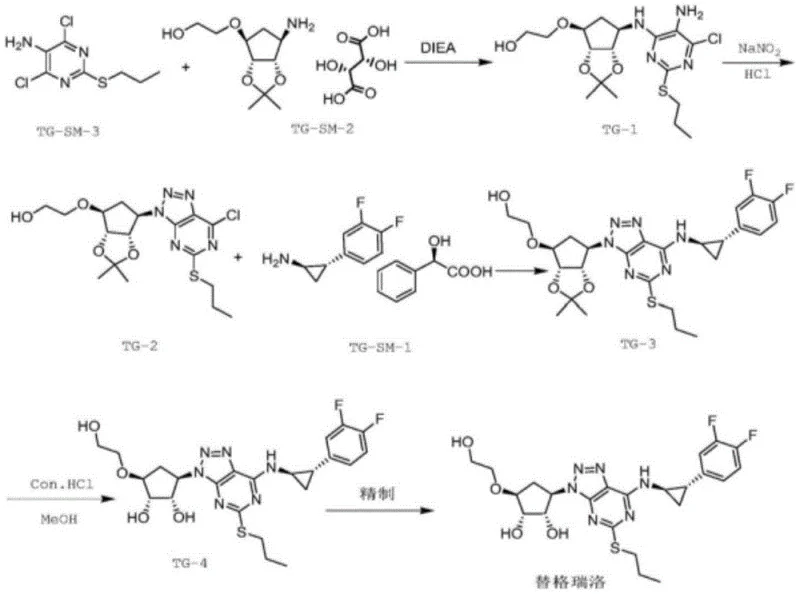
The Novel Approach
The innovative method disclosed in the patent overcomes these limitations by establishing a brand-new synthesis route specifically designed for Impurity VIII. This approach mainly comprises five strategic steps including condensation cyclization, substitution, and isopropylidene removal, all optimized for efficiency and purity. By adopting urea as a raw material and combining it with a specific condensing agent composition and alkaline reagent, the reaction proceeds under mild conditions that favor high product conversion. The process is designed to be simple to operate, making it highly suitable for large-scale industrial production of the Ticagrelor impurity compound VIII. This streamlined approach not only ensures the availability of high-purity reference standards but also significantly benefits the detection and monitoring of impurities throughout the Ticagrelor synthesis process, enhancing overall drug safety.
Mechanistic Insights into Urea-Mediated Condensation Cyclization
The core of this synthetic breakthrough lies in the initial condensation reaction where Compound I reacts with urea in the presence of a condensing agent composition. This step is critical for introducing the pyrimidine ring with a hydroxyl group, which is favorable for the subsequent substitution of 1-mercaptopropane and Compound IV. The use of a condensing agent composition such as EDCI, HOBT, and NMM facilitates the activation of the carboxyl group, allowing for efficient nucleophilic attack by the urea nitrogen. This mechanism ensures that the cyclization occurs with high regioselectivity, minimizing the formation of side products that could complicate downstream purification. The water-soluble nature of the by-products generated from this specific catalyst system further aids in their removal, contributing to the high purity of the intermediate Compound II. This mechanistic precision is essential for maintaining the structural integrity required for the final impurity standard.
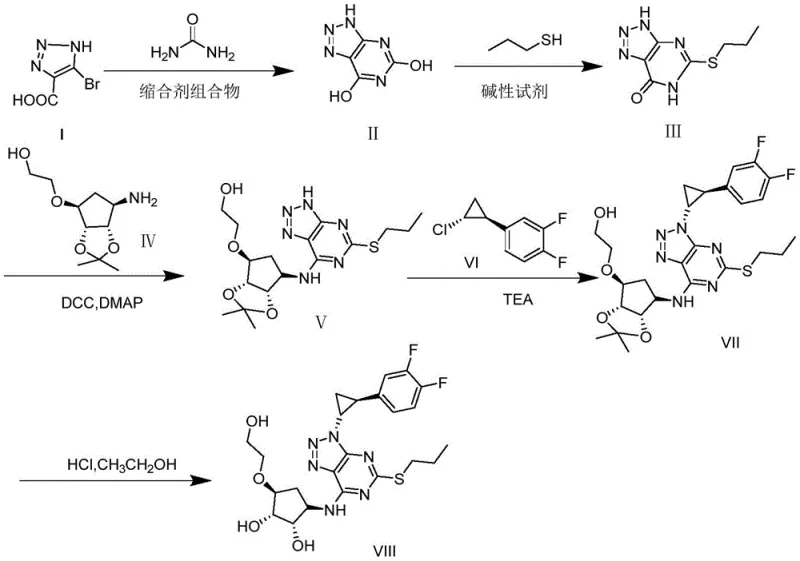
Impurity control is meticulously managed through the optimization of reaction conditions in subsequent steps, particularly during the substitution and deprotection phases. In the step involving the reaction with 1-mercaptopropane, the use of an alkaline reagent added in two portions helps to control the reaction rate and prevent the formation of thioether by-products through self-reaction. This careful addition strategy ensures that the nucleophilic substitution proceeds efficiently, yielding Compound III with high purity. Furthermore, the final deprotection step using concentrated hydrochloric acid in absolute ethanol is conducted at controlled temperatures to remove the isopropylidene group without degrading the sensitive triazolo-pyrimidine core. These mechanistic controls collectively ensure that the final Compound VIII meets the stringent purity requirements of 98% without the need for column chromatography, demonstrating a robust understanding of the chemical pathway.
How to Synthesize Ticagrelor Impurity VIII Efficiently
The synthesis of Ticagrelor Impurity VIII requires precise adherence to the optimized reaction parameters to ensure maximum yield and purity. The process begins with the condensation of Compound I and urea, followed by a series of substitution and coupling reactions that build the complex molecular architecture. Each step has been refined to operate under mild conditions, utilizing readily available solvents and reagents to facilitate ease of handling and scalability. The detailed standardized synthesis steps outlined below provide a clear roadmap for reproducing this high-quality impurity standard in a laboratory or industrial setting. Following these guidelines ensures that the critical quality attributes of the impurity are maintained, supporting accurate analytical method validation for Ticagrelor manufacturing.
- Condensation of Compound I with urea using EDCI/HOBT/NMM catalyst system to form the pyrimidine core.
- Substitution reaction with 1-mercaptopropane followed by coupling with the cyclopentyl amine derivative.
- Final deprotection and purification using hydrochloric acid in ethanol to achieve over 98% purity.
Commercial Advantages for Procurement and Supply Chain Teams
This novel synthesis route offers substantial commercial advantages by addressing key pain points in the supply chain for pharmaceutical intermediates. The elimination of complex purification steps like column chromatography drastically simplifies the production process, leading to significant cost savings in manufacturing operations. By using cheap and easily obtained raw materials such as urea, the dependency on expensive specialty reagents is reduced, enhancing the economic viability of large-scale production. This efficiency translates into a more reliable supply of high-purity reference standards, which is crucial for pharmaceutical companies maintaining strict quality control protocols. The robustness of the method ensures consistent batch-to-batch quality, reducing the risk of supply disruptions and supporting continuous manufacturing schedules.
- Cost Reduction in Manufacturing: The process achieves high yields and purity without the need for costly column chromatography purification, which significantly lowers the operational expenses associated with solvent consumption and resin costs. The use of economical raw materials like urea further drives down the cost of goods sold, making the production of this impurity standard more financially sustainable. By optimizing the reaction conditions to minimize by-product formation, the need for extensive rework or waste disposal is substantially reduced. These factors collectively contribute to a more cost-effective manufacturing process that aligns with the budget constraints of large-scale pharmaceutical production.
- Enhanced Supply Chain Reliability: The reliance on easily obtainable raw materials ensures that the supply chain is less vulnerable to shortages of specialty chemicals, thereby improving the continuity of supply. The simplicity of the operation and the mild reaction conditions reduce the technical barriers for manufacturing partners, allowing for a broader base of qualified suppliers. This flexibility enhances the resilience of the supply chain, ensuring that critical reference standards are available when needed for regulatory testing and quality assurance. The ability to scale this process efficiently means that demand spikes can be met without compromising on lead times or product quality.
- Scalability and Environmental Compliance: The method is designed for large-scale industrial production, with reaction conditions that are easily transferable from laboratory to pilot and commercial scales. The avoidance of hazardous reagents and the generation of water-soluble by-products simplify waste treatment processes, aligning with stringent environmental regulations. This eco-friendly approach reduces the environmental footprint of the manufacturing process, supporting corporate sustainability goals. The high conversion rate and yield minimize raw material waste, further contributing to a greener and more sustainable production model for pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Ticagrelor Impurity VIII. These answers are derived directly from the technical specifications and beneficial effects described in the patent data, ensuring accuracy and relevance for industry professionals. Understanding these details helps stakeholders make informed decisions about integrating this impurity standard into their quality control workflows. The information provided covers aspects of synthesis efficiency, purity standards, and scalability, which are critical for procurement and R&D planning.
Q: Why is synthesizing Ticagrelor Impurity VIII challenging?
A: Traditional methods often struggle with low yields and difficult separation due to the complex stereochemistry and similar polarity of byproducts, making high-purity reference standards hard to obtain without column chromatography.
Q: Does this new method require column chromatography?
A: No, the novel process described in patent CN110759917B achieves purity levels exceeding 98% through optimized crystallization and extraction steps, eliminating the need for costly and time-consuming column chromatography.
Q: Is this synthesis route scalable for industrial production?
A: Yes, the method utilizes cheap, easily obtained raw materials like urea and operates under mild conditions, making it highly suitable for large-scale industrial production and consistent supply chain reliability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ticagrelor Impurity VIII Supplier
The technical potential of this synthesis route underscores the importance of partnering with a CDMO expert capable of translating complex chemistry into commercial reality. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision. Our commitment to stringent purity specifications and the operation of rigorous QC labs guarantees that every batch of Ticagrelor Impurity VIII meets the highest industry standards. We understand the critical role that high-quality reference standards play in drug development and are dedicated to supporting your regulatory success through reliable supply and technical excellence.
We invite you to initiate a conversation about optimizing your supply chain for this critical intermediate. Our team is ready to provide a Customized Cost-Saving Analysis tailored to your specific production volumes and requirements. Please contact our technical procurement team to request specific COA data and route feasibility assessments that will demonstrate how we can support your project goals. Let us help you secure a stable and cost-effective supply of high-purity pharmaceutical intermediates for your global operations.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →