Advanced Synthesis of Lidocaine Impurity E for Commercial Scale-Up and Quality Control
Advanced Synthesis of Lidocaine Impurity E for Commercial Scale-Up and Quality Control
Introduction: Breaking New Ground in Impurity Standard Synthesis
The pharmaceutical industry relies heavily on the precise characterization of drug substances, necessitating the availability of high-purity impurity standards for rigorous quality control protocols. Patent CN111995539A introduces a groundbreaking preparation method for Lidocaine Hydrochloride Impurity E, also known chemically as 2,2'-azidobis(N-(2,6-dimethylphenyl)acetamide), which addresses the historical lack of efficient synthetic routes for this critical reference material. This innovation is particularly significant for manufacturers and quality assurance teams who require reliable access to certified standards to comply with stringent pharmacopoeial regulations, such as those outlined in the European Pharmacopoeia. By leveraging a novel nucleophilic substitution strategy, this technology enables the production of the target impurity with exceptional purity levels exceeding 98%, thereby reducing the analytical uncertainty in lidocaine batch testing. For a reliable pharmaceutical intermediates supplier, mastering this synthesis pathway represents a pivotal capability in supporting global drug safety and regulatory compliance initiatives.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the development of the technology disclosed in CN111995539A, the chemical community faced significant challenges in synthesizing Lidocaine Hydrochloride Impurity E with sufficient efficiency for commercial application. Existing literature and prior art lacked a dedicated, high-yield methodology, often forcing laboratories to rely on isolation from reaction by-products, which is inherently inefficient and unsustainable for scale-up. Comparative studies referenced within the patent data indicate that attempting to synthesize this dimeric structure using conventional ammonia-based conditions in solvents like 1,4-Dioxane or Toluene resulted in negligible conversion, yielding only trace amounts or no reaction at all even after extended reaction times of up to 48 hours. These failures highlight the kinetic barriers associated with forming the specific secondary amine linkage in this sterically hindered environment, where traditional reagents fail to drive the equilibrium toward the desired product. Consequently, the scarcity of this impurity standard has historically created bottlenecks in the supply chain for quality control laboratories, driving up costs and extending lead times for high-purity reference standards.
The Novel Approach
The novel approach detailed in the patent data overcomes these kinetic and thermodynamic barriers by employing a specific combination of activated acetamide derivatives and optimized acid-binding agents. Instead of relying on weak nucleophiles like ammonia, the process utilizes 2-(2,6-dimethylphenylamino)acetamide as a potent nucleophile to attack substituted N-(2,6-dimethylphenyl)-2-haloacetamide derivatives. This strategic selection of reactants ensures a much higher reactivity profile, allowing the reaction to proceed efficiently under moderate thermal conditions ranging from 25°C to 100°C. Furthermore, the method incorporates a staged heating protocol that initially facilitates mixing and initial bond formation at lower temperatures before ramping up to drive the reaction to completion, effectively minimizing the formation of side products. This results in a streamlined process that achieves yields between 88% and 90.7%, representing a drastic improvement over the trace results observed in conventional attempts and offering a viable path for cost reduction in API intermediate manufacturing.
Mechanistic Insights into Nucleophilic Substitution and Dimerization
The core chemical transformation driving this synthesis is a nucleophilic substitution reaction where the primary amine group of the starting acetamide derivative attacks the electrophilic carbon adjacent to the leaving group on the second acetamide molecule. 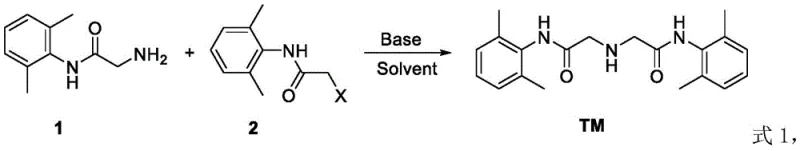 This mechanism is highly sensitive to the nature of the leaving group, denoted as X in the general formula, which can be a chloride, bromide, methoxy, or tosylate group. The patent data demonstrates that chloro- and bromo-derivatives serve as excellent electrophiles, facilitating rapid displacement by the amine nucleophile in the presence of a base. The choice of solvent plays a critical role in stabilizing the transition state; polar aprotic solvents like DMF and DMSO are shown to enhance the nucleophilicity of the amine by solvating the cation of the base without hydrogen bonding to the nucleophile itself. This solvation effect lowers the activation energy of the rate-determining step, ensuring that the reaction proceeds smoothly to form the central secondary amine linkage characteristic of Impurity E.
This mechanism is highly sensitive to the nature of the leaving group, denoted as X in the general formula, which can be a chloride, bromide, methoxy, or tosylate group. The patent data demonstrates that chloro- and bromo-derivatives serve as excellent electrophiles, facilitating rapid displacement by the amine nucleophile in the presence of a base. The choice of solvent plays a critical role in stabilizing the transition state; polar aprotic solvents like DMF and DMSO are shown to enhance the nucleophilicity of the amine by solvating the cation of the base without hydrogen bonding to the nucleophile itself. This solvation effect lowers the activation energy of the rate-determining step, ensuring that the reaction proceeds smoothly to form the central secondary amine linkage characteristic of Impurity E.
Impurity control is inherently built into this mechanistic pathway through the precise stoichiometry and the selection of the acid-binding agent. By maintaining a molar ratio of the nucleophile to the electrophile between 1:0.9 and 1:1.5, the process minimizes the risk of over-alkylation or polymerization, which are common pitfalls in amine acylation reactions. The use of organic bases such as diethylamine or N,N-diisopropylethylamine, or inorganic bases like sodium carbonate, effectively scavenges the acid by-product (HX) generated during the substitution. This neutralization prevents the protonation of the unreacted amine nucleophile, which would otherwise render it inactive and stall the reaction. The result is a clean reaction profile that yields the target dimer with purity levels consistently above 98%, significantly simplifying the downstream purification requirements and ensuring the material is suitable for use as a certified reference standard in analytical chromatography.
How to Synthesize Lidocaine Hydrochloride Impurity E Efficiently
Implementing this synthesis route in a production environment requires careful attention to the thermal profile and reagent addition sequence to maximize safety and yield. The process begins by dissolving the amine component in a selected solvent, followed by the controlled addition of the haloacetamide derivative and the base. 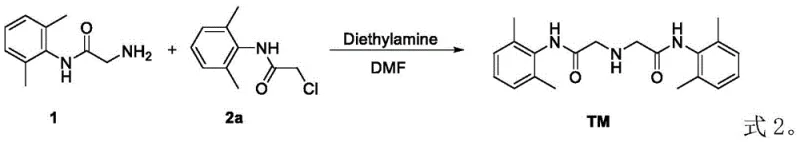 The reaction is then subjected to a multi-stage heating regimen, starting at ambient temperature to allow for initial mixing, followed by gradual heating to 60°C and finally to 90°C to ensure complete conversion. This specific thermal gradient is crucial for managing the exothermic nature of the substitution while driving the reaction to high conversion rates without degrading the product. Detailed standardized synthesis steps see the guide below.
The reaction is then subjected to a multi-stage heating regimen, starting at ambient temperature to allow for initial mixing, followed by gradual heating to 60°C and finally to 90°C to ensure complete conversion. This specific thermal gradient is crucial for managing the exothermic nature of the substitution while driving the reaction to high conversion rates without degrading the product. Detailed standardized synthesis steps see the guide below.
- Dissolve 2-(2,6-dimethylphenylamino)acetamide in a polar aprotic solvent such as DMF or DMSO under stirring.
- Add substituted N-(2,6-dimethylphenyl)-2-haloacetamide and an acid-binding agent like diethylamine or sodium carbonate.
- Heat the reaction mixture through staged temperature gradients from 25°C to 100°C to maximize yield and purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers substantial benefits for procurement managers and supply chain heads looking to optimize their sourcing strategies for pharmaceutical intermediates. The elimination of complex catalytic systems and the reliance on commodity chemicals such as DMF, diethylamine, and simple haloacetamides means that the raw material supply chain is robust and resistant to market volatility. This accessibility translates directly into enhanced supply chain reliability, as manufacturers are not dependent on scarce or expensive specialty reagents that could cause production delays. Furthermore, the high yield and purity achieved directly from the reaction workup reduce the need for extensive chromatographic purification, which is often a major cost driver in the production of fine chemicals. This efficiency allows for significant cost savings in manufacturing operations, making the final impurity standard more affordable for end-users without compromising on quality.
- Cost Reduction in Manufacturing: The process achieves high conversion efficiency without the need for expensive transition metal catalysts or complex purification infrastructure. By utilizing simple acid-binding agents and readily available solvents, the operational expenditure associated with reagent procurement and waste disposal is drastically simplified. The high yield reduces the amount of starting material required per unit of product, effectively lowering the cost of goods sold and allowing for more competitive pricing structures in the market for high-purity pharmaceutical intermediates.
- Enhanced Supply Chain Reliability: The reliance on bulk commodity chemicals ensures that production is not vulnerable to the supply constraints often associated with specialized reagents. The robustness of the reaction conditions, which tolerate a range of temperatures and solvents, provides manufacturing flexibility that can adapt to raw material availability. This flexibility ensures consistent delivery schedules and reduces the risk of stockouts, which is critical for laboratories that depend on a steady supply of reference standards for ongoing quality control testing and regulatory compliance.
- Scalability and Environmental Compliance: The reaction operates under moderate temperatures and atmospheric pressure, making it inherently safer and easier to scale from laboratory to commercial production volumes. The use of standard workup procedures involving water and ethyl acetate facilitates efficient solvent recovery and waste management, aligning with modern environmental compliance standards. This scalability ensures that the supply can grow in tandem with market demand, supporting the commercial scale-up of complex pharmaceutical intermediates without the need for specialized high-pressure or cryogenic equipment.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of Lidocaine Hydrochloride Impurity E. These insights are derived directly from the experimental data and beneficial effects described in the patent literature, providing a clear understanding of the technology's capabilities. Understanding these details helps stakeholders make informed decisions about integrating this material into their quality control workflows.
Q: What is the primary advantage of this synthesis method over prior art?
A: The method described in patent CN111995539A eliminates the need for complex purification steps, achieving purity levels exceeding 98% directly from the reaction workup, which significantly streamlines the production of reference standards.
Q: Which solvents are most effective for this nucleophilic substitution?
A: Polar aprotic solvents such as N,N-Dimethylformamide (DMF) and Dimethyl Sulfoxide (DMSO) have demonstrated superior solubility and reaction kinetics compared to non-polar alternatives like toluene, which failed to produce the target impurity in comparative studies.
Q: Is this process suitable for large-scale manufacturing of impurity standards?
A: Yes, the use of readily available industrial bases and moderate temperature ranges (25°C to 100°C) ensures that the process is safe and easily scalable for commercial production of high-purity pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Lidocaine Hydrochloride Impurity E Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity impurity standards play in ensuring the safety and efficacy of pharmaceutical products globally. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your volume requirements with consistent quality. We adhere to stringent purity specifications and utilize rigorous QC labs to verify that every batch of Lidocaine Hydrochloride Impurity E meets the necessary structural and purity criteria for analytical use. Our commitment to technical excellence ensures that you receive a product that supports accurate quantification and regulatory compliance.
We invite you to collaborate with us to optimize your supply chain for critical reference materials. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term quality control objectives.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →