Advanced Alpha-Carbon Alkylation of Mevinolin Analogs for Commercial Scale Production
Advanced Alpha-Carbon Alkylation of Mevinolin Analogs for Commercial Scale Production
The pharmaceutical industry continuously seeks more efficient pathways for synthesizing potent HMG-CoA reductase inhibitors, commonly known as statins. Patent CN1019395B discloses a groundbreaking method for the alpha-carbon alkylation of the 8-acyl group in mevinolin and its analogues, addressing critical limitations in prior art synthesis routes. This technology enables the direct transformation of naturally occurring fermentation products into highly active 2,2-dimethylbutyrate side chain derivatives through a streamlined process that significantly enhances overall yield and product purity. By utilizing a novel protection-alkylation-deprotection sequence, manufacturers can bypass the cumbersome multi-step procedures previously required, thereby optimizing the production of key pharmaceutical intermediates used in cholesterol-lowering therapies.
For R&D directors and process chemists, the significance of this patent lies in its ability to produce compounds with purity suitable for medical use through a simplified operational protocol. The method specifically targets the alpha-position of the acyl side chain, a modification known to increase biological activity compared to the natural alpha-methylbutyrate forms found in mevinolin and compactin. This technical advancement represents a pivotal shift in how high-purity statin intermediates are manufactured, offering a reliable statin intermediate supplier the capability to deliver superior quality materials with reduced process complexity and improved economic viability for large-scale operations.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of 2,2-dimethylbutyrate side chain analogs was plagued by inefficiency and low overall yields due to a convoluted four-step reaction sequence. As detailed in earlier patents such as U.S. Patent 4,444,784, the conventional route necessitated the initial removal of the natural 2-methylbutyrate side chain ester group, followed by the protection of the hydroxyl group on the pyrone ring. Subsequently, a re-esterification step was required to introduce the desired 2,2-dimethylbutyrate moiety, culminating in the removal of the 4-hydroxy protecting group. This lengthy pathway not only consumed excessive time and resources but also resulted in significant material loss at each stage, making it economically unattractive for commercial manufacturing environments where cost efficiency is paramount.
Furthermore, alternative methods disclosed in U.S. Patent 4,582,915 attempted to directly alkylate the alpha-carbon using metal alkylamides but suffered from severe drawbacks in drug commercialization. These processes required the repeated addition of amino base and methyl halide to achieve acceptable conversion rates, leading to time-consuming operations and moderate overall yields. A critical failure point was the necessity for selective hydrolysis to reduce unmethylated starting material to less than 0.7%, a step that could take up to 20 hours due to the slow hydrolysis kinetics of unconverted原料。Additionally, these methods generated a complex impurity profile, including debutylated mevinolin and double-methylated byproducts, pushing the final product purity to the borderline of acceptability for human health applications.
The Novel Approach
The method described in CN1019395B revolutionizes this landscape by introducing a streamlined protocol where only a single addition of alkali and alkyl halide is required to achieve high conversion and purity. This novel approach operates by first converting the starting lactone into an alkylamide and protecting the hydroxyl groups as bis-tert-butyldimethylsilyl ethers. This strategic protection allows for the selective alkylation at the alpha-position of the 8'-acyl side chain without affecting the sensitive lactone ring or other functional groups. The entire process of protection, carbon alkylation, and removal of the protecting group can be executed within the same reactor system, drastically reducing handling time and equipment requirements.
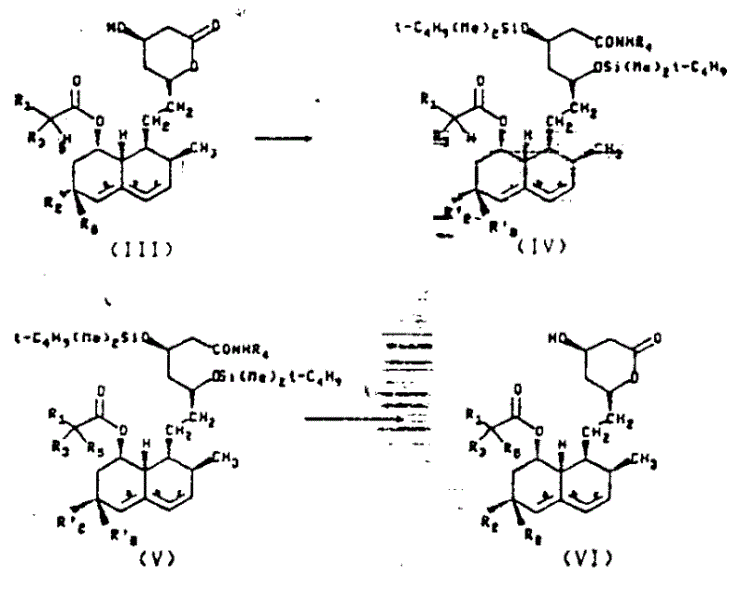
By employing this unified strategy, the invention eliminates the need for the tedious selective hydrolysis step that plagued previous methods, thereby ensuring that the number of impurities remains well below critical thresholds without the need for expensive repeated recrystallization. The result is a robust manufacturing process capable of producing batches with consistent quality and high purity, directly addressing the stability and reproducibility concerns that hindered earlier technologies. This efficiency translates directly into cost reduction in pharmaceutical intermediate manufacturing, as fewer unit operations and lower solvent consumption lead to a leaner, more sustainable production model.
Mechanistic Insights into Selective Alpha-Alkylation and Silyl Protection
The core of this technological breakthrough relies on the precise manipulation of reaction conditions to favor mono-alkylation at the specific alpha-carbon of the acyl side chain. The mechanism initiates with the formation of an amino alkali metal, preferably lithium tetrahydropyrrolide, generated by reacting n-butyllithium with a secondary amine in an anhydrous ether solvent like tetrahydrofuran at approximately -20°C. This strong base is then introduced to the cooled solution of the protected alkylamide at temperatures below -30°C, generating the enolate species necessary for nucleophilic attack. The subsequent single addition of an anhydrous alkyl halide, such as methyl iodide, ensures rapid and complete alkylation before any competing side reactions can occur, effectively locking in the desired 2,2-dimethyl substitution pattern.
Crucially, the use of tert-butyldimethylsilyl (TBS) protecting groups plays a vital role in controlling the impurity profile and preventing degradation of the sensitive beta-hydroxy lactone structure. By masking the hydroxyl groups at the 3 and 5 positions (or equivalent positions depending on the specific analog), the method prevents unwanted elimination reactions or ring-opening that typically occur under strongly basic conditions. Following the alkylation, the silyl groups are cleanly removed using aqueous hydrofluoric acid or similar reagents, and the alkylamide is smoothly converted back to the valerolactone form through controlled hydrolysis and heating. This mechanistic elegance ensures that the final product retains the stereochemical integrity required for biological activity while achieving the structural modification needed for enhanced potency.
How to Synthesize 2,2-Dimethylbutyrate Statins Efficiently
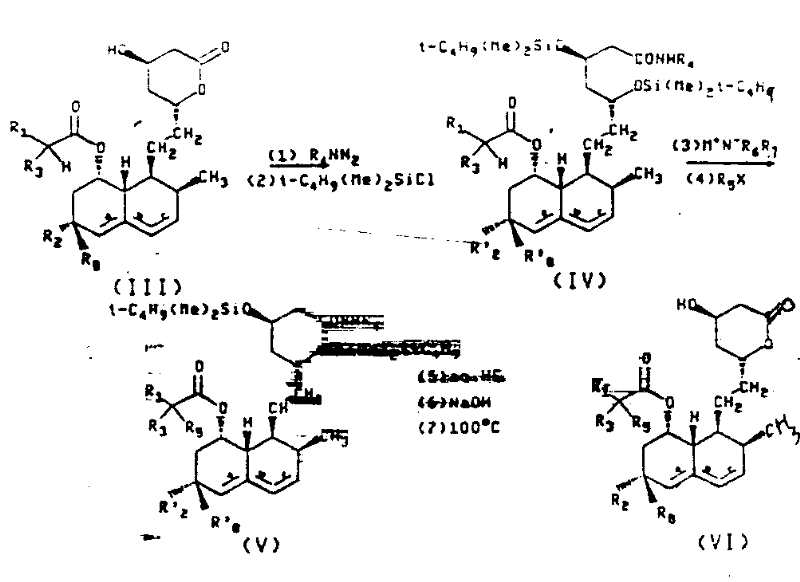
The synthesis of these high-value intermediates follows a logical progression designed to maximize yield while minimizing operational complexity. The process begins with the conversion of the raw material lactone, such as lovastatin, into a protected alkylamide derivative, setting the stage for the critical alkylation event. Detailed standardized synthetic steps see the guide below, which outlines the precise stoichiometry and thermal profiles required to replicate the patent's success in a pilot or production setting. Adhering to these parameters is essential for maintaining the delicate balance between reactivity and selectivity that defines this superior manufacturing route.
- Protect the hydroxyl groups of the starting lactone (e.g., Lovastatin) using tert-butyldimethylsilyl chloride and convert the lactone to an alkylamide.
- Perform selective alpha-alkylation by adding an alkali metal amide (e.g., lithium tetrahydropyrrolide) and an alkyl halide (e.g., methyl iodide) in a single addition sequence at low temperatures.
- Remove the silyl protecting groups and cyclize the alkylamide back to the lactone form through controlled hydrolysis and heating to obtain the high-purity final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented methodology offers substantial strategic benefits beyond mere technical superiority. The consolidation of multiple reaction steps into a streamlined sequence significantly reduces the total processing time and the volume of solvents and reagents required per kilogram of finished product. This reduction in material intensity directly correlates to a lower cost of goods sold (COGS), allowing suppliers to offer more competitive pricing structures without compromising on margin. Furthermore, the elimination of difficult-to-control steps like selective hydrolysis reduces the risk of batch failures, ensuring a more reliable supply of critical intermediates for downstream API synthesis.
- Cost Reduction in Manufacturing: The primary driver for cost optimization in this process is the drastic simplification of the synthetic route, which removes the need for intermediate isolation and purification steps associated with the older four-step methods. By avoiding the repeated addition of expensive reagents and the extended reaction times required for slow hydrolysis, manufacturers can achieve significant operational savings. Additionally, the higher overall yield means that less starting material is wasted, further enhancing the economic efficiency of producing high-purity HMG-CoA reductase inhibitors on a commercial scale.
- Enhanced Supply Chain Reliability: The robustness of this single-addition alkylation method contributes to greater supply chain stability by minimizing the variables that can lead to production delays. Since the process does not rely on the slow and variable kinetics of selective hydrolysis, cycle times are more predictable, allowing for better production planning and inventory management. The use of readily available starting materials like lovastatin and compactin, combined with common industrial reagents, ensures that sourcing bottlenecks are minimized, securing a continuous flow of materials for global pharmaceutical customers.
- Scalability and Environmental Compliance: From an environmental and scalability perspective, the reduction in solvent usage and waste generation aligns with modern green chemistry principles and regulatory expectations. The ability to perform protection, alkylation, and deprotection in the same reactor vessel reduces the equipment footprint and energy consumption associated with transferring materials between different processing units. This streamlined approach facilitates the commercial scale-up of complex lipid-lowering agents, enabling manufacturers to meet increasing global demand while adhering to strict environmental discharge standards and safety protocols.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this alpha-alkylation technology. These insights are derived directly from the patent specifications and are intended to clarify the operational advantages and chemical nuances of the process for potential partners and stakeholders. Understanding these details is crucial for evaluating the feasibility of integrating this method into existing manufacturing portfolios.
Q: How does this alkylation method improve purity compared to prior art?
A: Unlike previous methods requiring four distinct chemical steps including selective hydrolysis which often left unmethylated starting material, this novel approach utilizes a single addition of base and alkyl halide. This minimizes the formation of impurities such as debutylated mevinolin and double-methylated byproducts, ensuring the final product meets stringent pharmaceutical purity specifications without extensive recrystallization.
Q: What are the critical temperature controls for this synthesis?
A: The process requires precise thermal management, specifically cooling the protected alkylamide solution to approximately -35°C before adding the amino alkali metal. The alkylation step must be maintained below -30°C during addition, followed by a controlled warm-up to -10°C. Final lactonization involves heating the ammonium salt suspension to 100°C, demonstrating a robust yet temperature-sensitive profile suitable for industrial reactors.
Q: Can this process be scaled for commercial manufacturing of HMG-CoA inhibitors?
A: Yes, the method is designed for scalability by eliminating the need for repeated additions of reagents and complex isolation steps found in older patents. The use of common solvents like tetrahydrofuran and cyclohexane, along with standard reagents like n-butyllithium and silyl chlorides, facilitates the commercial scale-up of complex lipid-lowering agents while maintaining consistent batch-to-batch quality.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Mevinolin Analog Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced synthetic routes like the one described in CN1019395B for the production of next-generation statin intermediates. As a dedicated CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that complex chemical transformations are executed with precision and consistency. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of high-purity HMG-CoA reductase inhibitors meets the exacting standards required by the global pharmaceutical industry.
We invite forward-thinking organizations to collaborate with us to leverage these technological advancements for their supply chain optimization. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to obtain specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in commercial scale-up of complex polymer additives and pharmaceutical intermediates can drive value and efficiency for your organization.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →