Advanced Manufacturing of High-Purity Citalopram HBr via Crystalline Salt Isolation
The pharmaceutical industry continuously demands higher standards for Active Pharmaceutical Ingredients (APIs), particularly for widely prescribed antidepressants like Citalopram. Patent CN1556800A introduces a groundbreaking methodology that addresses the longstanding challenges associated with achieving pharmaceutical-grade purity, specifically targeting a purity level exceeding 99.7%. This technical insight report analyzes the novel process for preparing Citalopram Hydrobromide (HBr), focusing on the strategic isolation of crystalline intermediates and advanced purification techniques. Unlike conventional methods that struggle with oily, unpurifiable intermediates, this invention leverages the formation of a stable crystalline salt early in the synthesis pathway. This fundamental shift not only enhances the chemical integrity of the final product but also streamlines the manufacturing workflow, offering a robust solution for reliable pharmaceutical intermediate supplier networks seeking to optimize their production capabilities.
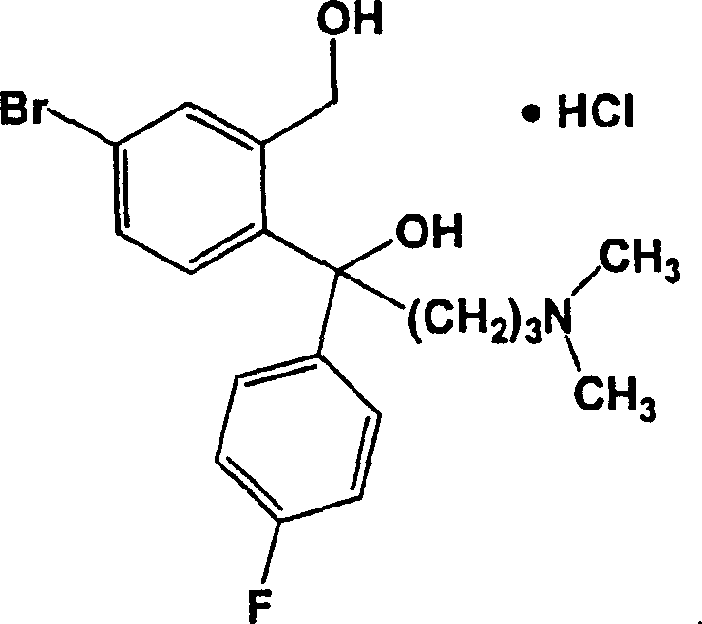
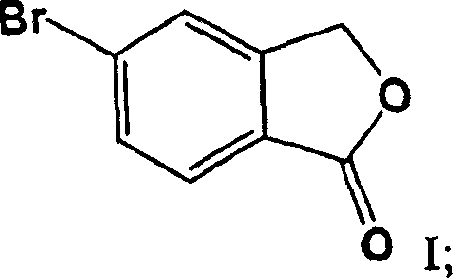
For R&D directors and process chemists, understanding the limitations of historical synthesis routes is crucial for evaluating new technologies. The conventional method, originally disclosed in US Patent 4,136,193, relies on a five-step conversion starting from 5-bromobenzo[c]furanone. A critical bottleneck in this legacy process is the formation of the bromodiol intermediate (Formula III) as a free amine oil. Because oils cannot be effectively purified through crystallization or recrystallization, impurities generated in early steps carry through to the final product, making it nearly impossible to meet stringent quality standards without complex downstream processing. Furthermore, the conventional route necessitates the use of hazardous solvents like diethyl ether and benzene, posing significant safety and environmental compliance risks during commercial scale-up of complex pharmaceutical intermediates.
In stark contrast, the novel approach detailed in CN1556800A revolutionizes the synthesis by converting the oily intermediate into a crystalline hydrochloride salt immediately after the Grignard reactions. This transformation allows for rigorous purification via recrystallization, effectively stripping away impurities before the molecule undergoes cyclization and cyanation. Additionally, the new process optimizes the cyclization step by drastically reducing the amount of phosphoric acid required. While the prior art demanded a massive excess of approximately 30 equivalents of 60% phosphoric acid, the new method achieves efficient ring closure with only 2 to 10 equivalents. This reduction not only simplifies the neutralization workup but also mitigates the severe exothermic risks associated with neutralizing large volumes of strong acid, thereby enhancing operational safety and cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Crystalline Salt Isolation and Impurity Control
The core mechanistic advantage of this invention lies in the manipulation of the intermediate's physical state to facilitate purification. The process begins with the reaction of 5-bromobenzo[c]furanone with 4-fluorophenylmagnesium bromide, followed by the addition of dimethylaminopropylmagnesium chloride. In traditional syntheses, quenching this mixture yields an oil. However, by quenching with an acid such as aqueous HCl, the process forces the precipitation of the Formula III compound as a crystalline hydrochloride salt. This solid form possesses a defined melting point (approximately 183°C) and can be washed and recrystallized from solvents like 2-butanol. This step acts as a 'purity gate,' ensuring that only high-quality material proceeds to the cyclization stage, which is essential for producing high-purity OLED material or pharmaceutical grades where trace impurities are unacceptable.
Following the isolation of the pure crystalline salt, the synthesis proceeds to cyclization and cyanation. A notable mechanistic refinement occurs in the cyanation step, where a mixture of cuprous cyanide and sodium cyanide is employed instead of cuprous cyanide alone. This modification significantly improves reaction kinetics and conversion rates compared to the sluggish performance of the prior art. Furthermore, the patent describes a sophisticated impurity removal strategy for the final product. Demethylated and dedimethylated impurities, which are structurally similar to Citalopram and difficult to separate via standard extraction, are effectively removed using a scavenger resin. This resin contains functional groups, such as isocyanates, that react irreversibly with the primary and secondary amines present in the impurities but do not react with the tertiary amine of the desired Citalopram product. This selective chemical binding renders the impurities insoluble, allowing for their removal via simple filtration.
How to Synthesize Citalopram HBr Efficiently
The synthesis of Citalopram HBr via this novel route involves a sequence of precise chemical transformations designed to maximize yield and purity. The process initiates with the formation of the Grignard reagents and their subsequent reaction with the phthalide starting material under controlled temperatures to prevent side reactions. The critical quenching step with hydrochloric acid must be managed carefully to ensure the formation of the correct crystalline polymorph. Following isolation, the salt undergoes acid-catalyzed cyclization in an organic solvent like toluene, followed by a cyanation reaction using the optimized copper/sodium cyanide mixture. The final stages involve the formation of the hydrobromide salt and optional recrystallization to achieve the highest pharmaceutical standards. For detailed operational parameters, stoichiometry, and safety protocols, refer to the standardized synthesis guide below.
- React 5-bromobenzo[c]furanone with 4-fluorophenylmagnesium bromide and dimethylaminopropylmagnesium chloride, then quench with aqueous HCl to isolate the crystalline Formula III HCl salt.
- Perform ring closure on the crystalline salt using reduced equivalents (2-10 eq) of phosphoric acid in toluene, followed by neutralization and extraction.
- Execute cyanation using a CuCN/NaCN mixture, followed by Simulated Moving Bed (SMB) chromatography and scavenger resin treatment to remove trace impurities.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of the CN1556800A process offers substantial strategic benefits that extend beyond mere chemical efficiency. The ability to isolate and purify intermediates as crystalline solids significantly de-risks the supply chain by ensuring consistent quality batch-to-batch. In the context of reducing lead time for high-purity pharmaceutical intermediates, this process eliminates the need for extensive and often unpredictable downstream purification efforts that are characteristic of oil-based intermediate routes. By establishing purity early in the synthesis, manufacturers can reduce cycle times and minimize the risk of batch failures, which is a critical factor for maintaining supply continuity in the global market.
- Cost Reduction in Manufacturing: The novel process delivers significant cost savings through the optimization of reagent consumption and waste management. By reducing the phosphoric acid load from 30 equivalents to less than 10 equivalents, the process drastically lowers the cost of raw materials and the associated costs of neutralizing and disposing of acidic waste streams. Furthermore, the replacement of hazardous solvents like benzene and diethyl ether with safer alternatives like toluene reduces the regulatory burden and insurance costs associated with handling highly flammable and toxic substances. These efficiencies collectively contribute to a more economically viable production model without compromising on quality.
- Enhanced Supply Chain Reliability: The stability of the crystalline intermediate provides a buffer against supply chain disruptions. Unlike oily intermediates which may degrade or vary in quality during storage, the crystalline HCl salt of Formula III is stable and can be stockpiled if necessary. This flexibility allows manufacturers to decouple production stages, optimizing equipment utilization and ensuring that delays in one part of the process do not halt the entire production line. This robustness is essential for partners seeking a reliable pharmaceutical intermediate supplier capable of meeting demanding delivery schedules.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, addressing the thermal hazards that often limit batch sizes in conventional synthesis. The reduction in exothermic potential during the neutralization of the cyclization mixture allows for larger batch sizes and safer operation in standard reactor vessels. Additionally, the implementation of scavenger resin technology for impurity removal minimizes solvent usage compared to multiple recrystallization or chromatography steps, aligning with modern green chemistry principles and reducing the environmental footprint of the manufacturing process.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this advanced synthesis route. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the practical application of the technology. Understanding these details is vital for technical teams evaluating the feasibility of integrating this process into existing manufacturing frameworks.
Q: How does the new process improve purity compared to US Patent 4,136,193?
A: The novel process isolates the intermediate (Formula III) as a crystalline HCl salt rather than an oil, allowing for recrystallization and impurity removal before the final steps, ensuring >99.7% purity.
Q: What are the safety advantages of the new cyclization method?
A: The new method reduces phosphoric acid usage from 30 equivalents to approximately 2-10 equivalents, significantly minimizing the exothermic heat generated during neutralization and reducing waste disposal costs.
Q: How are demethylated impurities removed in this synthesis?
A: The process utilizes a scavenger resin with functional groups reactive to primary and secondary amines, which selectively binds demethylated impurities while leaving the tertiary amine product unaffected.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Citalopram HBr Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to meet the evolving needs of the global pharmaceutical market. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovations like the CN1556800A process can be successfully translated from the laboratory to full-scale manufacturing. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of Citalopram HBr meets the highest international standards. Our infrastructure is designed to handle complex chemistries safely and efficiently, providing our partners with a secure and dependable source of high-quality intermediates.
We invite you to collaborate with us to leverage these technological advancements for your supply chain. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your project goals. Let us partner with you to deliver superior value through scientific excellence and operational reliability.