Advanced Synthetic Route for Oral Carbapenem Intermediates Enhancing Manufacturing Efficiency
Advanced Synthetic Route for Oral Carbapenem Intermediates Enhancing Manufacturing Efficiency
The pharmaceutical industry continuously seeks robust methodologies for producing high-value antibiotic intermediates, particularly for the 1β-methylcarbapenem class which exhibits superior in vivo stability and broad-spectrum antibacterial activity. Patent CN1708504A discloses a groundbreaking approach to synthesizing novel β-lactam compounds that serve as critical co-synthetic intermediates for oral administration drugs. This technology addresses longstanding inefficiencies in traditional manufacturing pathways by introducing a strategic protection-deprotection sequence that utilizes specific silyl groups to enhance reaction fidelity. By shifting the introduction of complex thiol residues to the final stages of the synthesis, this method significantly mitigates the economic risks associated with early-stage yield losses. For R&D directors and procurement specialists, understanding this mechanistic shift is vital for evaluating potential supply chain optimizations and cost structures in API manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the production of 1β-methylcarbapenem compounds for oral administration has been plagued by inefficient multi-step sequences that necessitate early introduction of expensive thiol reagents. As illustrated in prior art methods, such as those converting compound (7) to (8) and subsequently to (9), manufacturers were forced to carry costly sulfur-containing moieties through multiple protection and deprotection cycles. 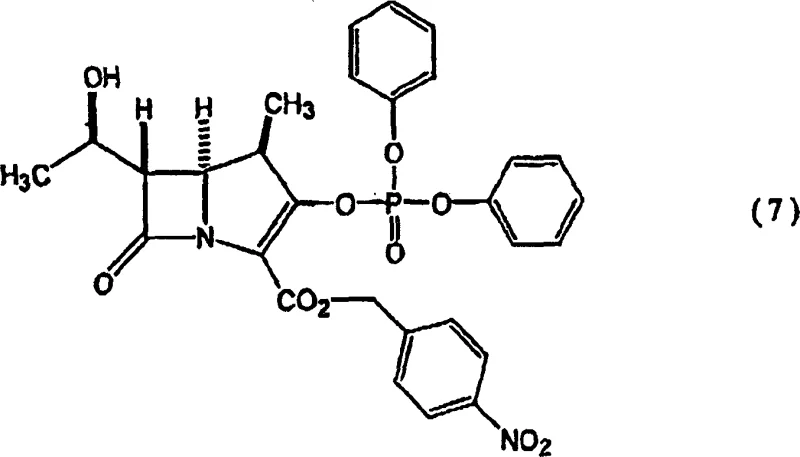 This traditional approach not only inflated raw material costs but also complicated purification processes due to the sensitivity of the thioester functionality under various reaction conditions. Furthermore, the requirement to convert carboxylic acid protecting groups repeatedly added unnecessary operational complexity, leading to lower overall throughput and increased waste generation. The reliance on harsh deprotection conditions for bulky silyl groups, such as tert-butyldimethylsilyl found in compound (17), often resulted in the degradation of the sensitive β-lactam core, thereby compromising the purity profile required for pharmaceutical grade intermediates.
This traditional approach not only inflated raw material costs but also complicated purification processes due to the sensitivity of the thioester functionality under various reaction conditions. Furthermore, the requirement to convert carboxylic acid protecting groups repeatedly added unnecessary operational complexity, leading to lower overall throughput and increased waste generation. The reliance on harsh deprotection conditions for bulky silyl groups, such as tert-butyldimethylsilyl found in compound (17), often resulted in the degradation of the sensitive β-lactam core, thereby compromising the purity profile required for pharmaceutical grade intermediates.
The Novel Approach
The innovative methodology presented in this patent fundamentally restructures the synthetic timeline by deferring the coupling of the thiol residue until the final transformation steps. Instead of early thiolation, the process focuses on the efficient construction of a versatile phosphorylated intermediate, specifically compound (3), which acts as a universal precursor. 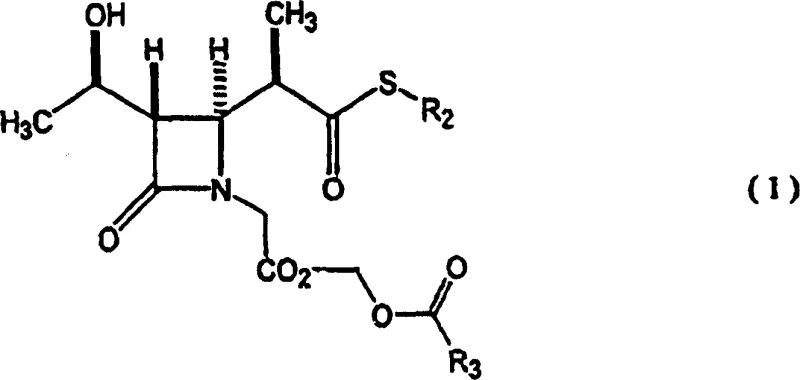 This novel route involves the initial protection of the hydroxyl group with a trimethylsilyl or triethylsilyl moiety, followed by a concerted cyclization and phosphorylation sequence. By utilizing diphenyl chlorophosphate, the method creates a highly reactive enol phosphate species that is primed for nucleophilic attack by various thiols in a single subsequent step. This strategic decoupling of the carbon skeleton construction from the side-chain installation allows for greater flexibility in producing diverse carbapenem derivatives from a common, stable intermediate, thereby streamlining inventory management and reducing the complexity of the manufacturing workflow.
This novel route involves the initial protection of the hydroxyl group with a trimethylsilyl or triethylsilyl moiety, followed by a concerted cyclization and phosphorylation sequence. By utilizing diphenyl chlorophosphate, the method creates a highly reactive enol phosphate species that is primed for nucleophilic attack by various thiols in a single subsequent step. This strategic decoupling of the carbon skeleton construction from the side-chain installation allows for greater flexibility in producing diverse carbapenem derivatives from a common, stable intermediate, thereby streamlining inventory management and reducing the complexity of the manufacturing workflow.
Mechanistic Insights into Silyl Protection and Phosphorylation Cyclization
The core chemical innovation lies in the specific selection of the silyl protecting group and the subsequent base-mediated cyclization mechanism. The patent specifies the use of trimethylsilyl (TMS) or triethylsilyl (TES) groups, which offer a distinct advantage over bulkier alternatives due to their lability under mild acidic conditions. During the deprotection phase, these groups can be cleaved at a pH range of 2 to 6 using agents like citric acid or dilute hydrochloric acid, ensuring that the fragile β-lactam ring and the newly formed double bond remain intact. 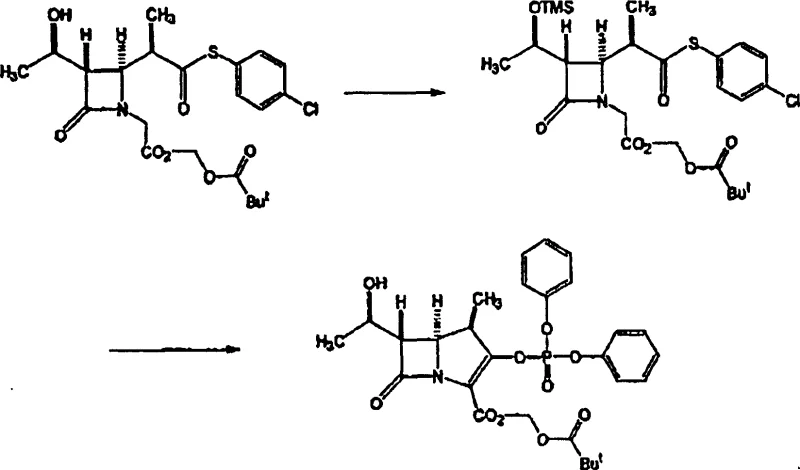 This contrasts sharply with prior art methods requiring fluoride sources or stronger acids that could induce ring opening or epimerization. The cyclization step itself is driven by strong bases such as potassium tert-butoxide or sodium hydride, which abstract the acidic proton adjacent to the carbonyl, facilitating intramolecular ring closure to form the bicyclic system. The immediate trapping of the resulting enolate with diphenyl chlorophosphate prevents retro-aldol reactions and stabilizes the intermediate as a phosphate ester, which serves as an excellent leaving group for the final thiol substitution.
This contrasts sharply with prior art methods requiring fluoride sources or stronger acids that could induce ring opening or epimerization. The cyclization step itself is driven by strong bases such as potassium tert-butoxide or sodium hydride, which abstract the acidic proton adjacent to the carbonyl, facilitating intramolecular ring closure to form the bicyclic system. The immediate trapping of the resulting enolate with diphenyl chlorophosphate prevents retro-aldol reactions and stabilizes the intermediate as a phosphate ester, which serves as an excellent leaving group for the final thiol substitution.
Impurity control is inherently built into this mechanism through the use of scavengers during the cyclization phase. The reaction generates metal thiolate by-products which, if left unchecked, could interfere with the phosphorylation or subsequent steps. The protocol recommends the addition of alkylating agents like methyl iodide or benzyl bromide, or even the diphenyl chlorophosphate itself in excess, to capture these metal thiolates effectively. This ensures that the final crude product contains minimal sulfur-based impurities, simplifying downstream crystallization and purification. Furthermore, the choice of solvents such as tetrahydrofuran or toluene mixtures optimizes the solubility of both the ionic bases and the organic substrates, promoting homogeneous reaction kinetics that are essential for consistent batch-to-batch reproducibility in a commercial setting.
How to Synthesize 1β-Methylcarbapenem Intermediates Efficiently
The synthesis of these high-value intermediates requires precise control over reaction parameters to maximize yield and stereochemical integrity. The process begins with the silylation of the hydroxyl group on the azetidinone precursor, followed by a low-temperature cyclization that demands careful thermal management to prevent decomposition. 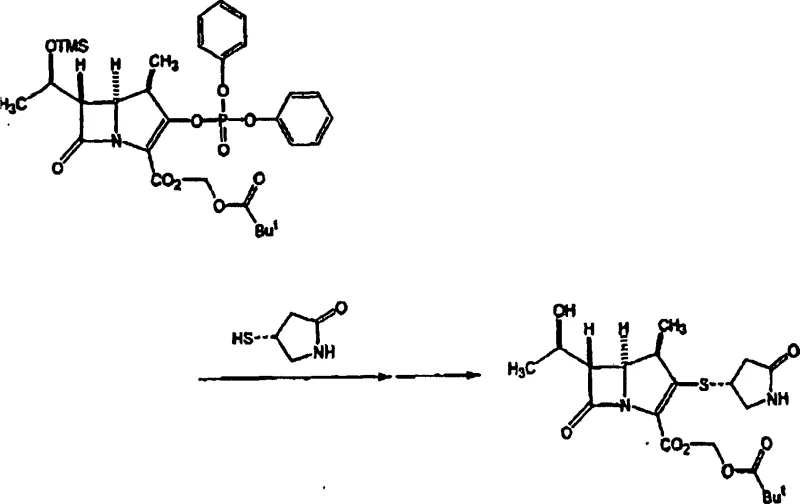 The subsequent phosphorylation and final thiol coupling steps are designed to be telescoped where possible, reducing the number of isolation events and solvent exchanges. Detailed standardized operating procedures for scaling this chemistry from laboratory to pilot plant are critical for maintaining the delicate balance between reaction rate and selectivity. For a comprehensive breakdown of the specific molar ratios, temperature profiles, and workup procedures validated in the patent examples, please refer to the technical guide below.
The subsequent phosphorylation and final thiol coupling steps are designed to be telescoped where possible, reducing the number of isolation events and solvent exchanges. Detailed standardized operating procedures for scaling this chemistry from laboratory to pilot plant are critical for maintaining the delicate balance between reaction rate and selectivity. For a comprehensive breakdown of the specific molar ratios, temperature profiles, and workup procedures validated in the patent examples, please refer to the technical guide below.
- Protect the hydroxyl group of the starting β-lactam compound using trimethylsilyl or triethylsilyl chloride in the presence of an amine base.
- Perform cyclization using a strong base like potassium tert-butoxide, followed by reaction with diphenyl chlorophosphate to form the phosphorylated intermediate.
- Execute mild acidic deprotection to remove the silyl group, enabling subsequent one-step thiol introduction for the final carbapenem structure.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers substantial advantages by decoupling the production of the core scaffold from the sourcing of specialized thiol reagents. Traditional methods required purchasing expensive, often custom-synthesized thiols at the beginning of the process, tying up capital in materials that would undergo multiple transformative steps with inherent yield attrition. By contrast, this novel approach allows manufacturers to stockpile the stable, phosphorylated intermediate (compound 3 or 4) and only introduce the specific thiol residue when demand for a particular final API is confirmed. This just-in-time manufacturing capability drastically reduces inventory carrying costs and minimizes the risk of obsolescence for specialized raw materials. Additionally, the use of commodity chemicals for the protection and cyclization steps, such as trimethylsilyl chloride and diphenyl chlorophosphate, ensures a robust and competitive supply chain for the bulk of the synthesis.
- Cost Reduction in Manufacturing: The elimination of early-stage thiol introduction removes the need to process high-value sulfur compounds through multiple purification cycles, significantly lowering the cost of goods sold. By deferring the addition of these expensive reagents to the final step, the overall material efficiency is improved because yield losses in earlier steps do not result in the loss of the costly thiol moiety. Furthermore, the mild deprotection conditions reduce the consumption of specialized reagents and minimize waste treatment costs associated with hazardous by-products, contributing to a leaner and more economically viable production model.
- Enhanced Supply Chain Reliability: Utilizing a common intermediate for multiple carbapenem derivatives enhances supply chain resilience by allowing for flexible production scheduling. Manufacturers can produce large batches of the universal precursor without committing to a specific final drug product, thereby buffering against fluctuations in market demand for individual antibiotics. The reliance on widely available silylating agents and phosphorus reagents rather than niche catalysts also mitigates the risk of supply disruptions, ensuring continuous availability of critical intermediates for downstream API synthesis.
- Scalability and Environmental Compliance: The process is designed for scalability, utilizing solvents and reagents that are compatible with standard industrial reactor setups and safety protocols. The ability to perform deprotection under mild acidic conditions reduces the corrosive load on equipment and lowers the environmental impact of waste streams compared to processes requiring harsh fluorides or strong mineral acids. This alignment with green chemistry principles facilitates easier regulatory approval and supports sustainability goals, making the technology attractive for long-term commercial partnerships focused on responsible manufacturing practices.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic pathway. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on reaction scope and limitations. Understanding these nuances is essential for process chemists evaluating the feasibility of adopting this route for commercial production.
Q: Why is the trimethylsilyl protecting group preferred over tert-butyldimethylsilyl in this synthesis?
A: The trimethylsilyl group allows for deprotection under significantly milder acidic conditions (pH 2-6) compared to bulkier silyl groups, minimizing the risk of decomposing sensitive β-lactam functional groups during the final stages of synthesis.
Q: What represents the primary cost advantage of this novel synthetic route?
A: The primary economic benefit stems from delaying the introduction of expensive thiol residues until the final synthetic steps, thereby reducing the financial risk associated with yield losses in earlier multi-stage transformations.
Q: Which solvents are optimal for the cyclization and phosphorylation reactions?
A: Tetrahydrofuran (THF) or mixtures of THF and toluene are identified as optimal solvents due to their ability to dissolve both the strong bases and the organic intermediates while maintaining stability at low reaction temperatures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1β-Methylcarbapenem Intermediate Supplier
The technological advancements detailed in patent CN1708504A represent a significant leap forward in the efficient production of oral carbapenem intermediates, offering a pathway to higher purity and reduced manufacturing complexity. NINGBO INNO PHARMCHEM stands at the forefront of translating such innovative chemical processes into commercial reality, leveraging our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs and adheres to stringent purity specifications, ensuring that every batch of intermediate meets the exacting standards required for global pharmaceutical supply chains. We understand the critical nature of antibiotic intermediates and are committed to delivering consistent quality that supports your regulatory filings and market launch timelines.
We invite you to collaborate with our technical team to explore how this optimized synthesis can be integrated into your existing supply network. By partnering with us, you gain access to a Customized Cost-Saving Analysis that quantifies the potential economic benefits of switching to this novel route for your specific portfolio. Please contact our technical procurement team today to request specific COA data and route feasibility assessments tailored to your project requirements, and let us demonstrate how our expertise can drive value and efficiency in your carbapenem manufacturing operations.