Advanced Synthesis of 1-Methyl Carbacephem Intermediates for Commercial Antibiotic Production
The pharmaceutical industry constantly seeks more efficient pathways for producing beta-lactam antibiotics, and patent CN101054376A presents a significant breakthrough in the synthesis of carbacephem intermediates. This document details a novel method for preparing (6R, 7S)-7-(1R-hydroxyethyl)-4-allyl carboxylate-3-substituted-3-ene-1-methyl carbacephem, a critical scaffold for next-generation antibacterial agents. Unlike traditional approaches that often rely on complex and hazardous diazonium chemistry, this invention leverages a streamlined sequence starting from a protected azetidinone. The process integrates a Wittig-Horner olefination followed by a unique triethyl phosphite-mediated cyclization, offering a safer and more controllable alternative for high-purity pharmaceutical intermediate production. By redefining the construction of the cephem nucleus, this technology addresses key pain points in impurity control and operational safety.
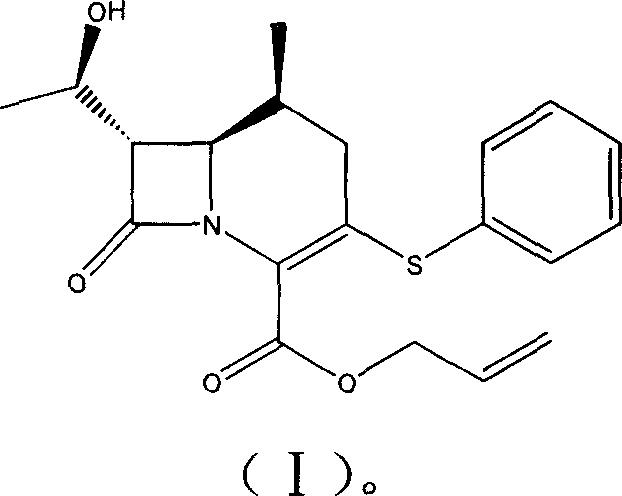
For procurement managers and supply chain directors, understanding the structural integrity of the final product is essential for quality assurance. The target molecule, designated as Compound (I), features a fused four-and-six-membered ring system characteristic of carbacephems, with specific stereochemistry at the 6R and 7S positions. The presence of the allyl ester at the 4-position and the hydroxyethyl side chain at the 7-position are critical for subsequent biological activity and further derivatization. Securing a reliable pharmaceutical intermediate supplier who can consistently deliver this specific stereoisomer is vital for maintaining the efficacy of the final antibiotic drug product. The structural complexity necessitates a robust synthetic route that minimizes epimerization and ensures high optical purity throughout the manufacturing campaign.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of carbacephem cores, as disclosed in earlier patents like US4174316, has relied heavily on L-aspartic acid as the starting chiral pool material. This conventional pathway involves converting the amino acid into a 4-iodomethyl-2-carbonyl azetidine intermediate, followed by a carbon extension reaction. The most significant bottleneck in this traditional route is the ring-closure step, which typically utilizes diazonium salts to merge the six-membered ring onto the beta-lactam nucleus. Diazonium chemistry is notoriously unstable, posing severe safety risks due to potential explosivity and requiring stringent temperature controls that complicate commercial scale-up of complex pharmaceutical intermediates. Furthermore, the handling of heavy metal catalysts often required in these older sequences introduces additional costs for metal scavenging and waste treatment, negatively impacting the overall cost structure of antibiotic manufacturing.
The Novel Approach
In stark contrast, the methodology described in CN101054376A circumvents these hazards by employing a completely different strategic disconnection. The new route initiates with a Wittig-Horner reaction on a pre-formed azetidinone, establishing the necessary carbon framework without the need for unstable diazonium species. The pivotal innovation lies in the ring-closing step, which utilizes triethyl phosphite and a catalytic amount of hydroquinone to effect "carbonyl docking." This phosphite-mediated cyclization is not only safer but also operates under reflux conditions in xylene, which is far more amenable to standard industrial reactor setups than cryogenic diazonium protocols. This shift represents a substantial advancement in cost reduction in pharmaceutical intermediate manufacturing, as it eliminates the need for specialized safety infrastructure and reduces the burden of hazardous waste disposal, thereby enhancing the overall economic viability of the production process.
Mechanistic Insights into Triethyl Phosphite-Mediated Cyclization
The core of this synthetic strategy revolves around the precise construction of the cephem ring system through a series of well-defined transformations. The process begins with the reaction of (3S, 4S)-3-[1-(R)-tert-butyldimethylsiloxyethyl]-4-[(S)-acetyl]azetidin-2-one with ethyl diethoxyphosphonoacetate in the presence of sodium hydride. This Wittig-Horner olefination generates a mixture of trans (III) and cis (IV) isomers, which are subsequently subjected to catalytic hydrogenation using Pd/C or PtO2. This reduction step is crucial as it saturates the double bond and establishes the correct stereochemistry at the side chain, yielding the ethyl ester intermediate (V). Following hydrolysis with potassium hydroxide, the resulting acid (VI) is activated using N,N'-carbonyldiimidazole (CDI) and coupled with a substituted thiol to form the thioester (VII). This thioester functionality is key, as it serves as the leaving group precursor for the subsequent cyclization event.
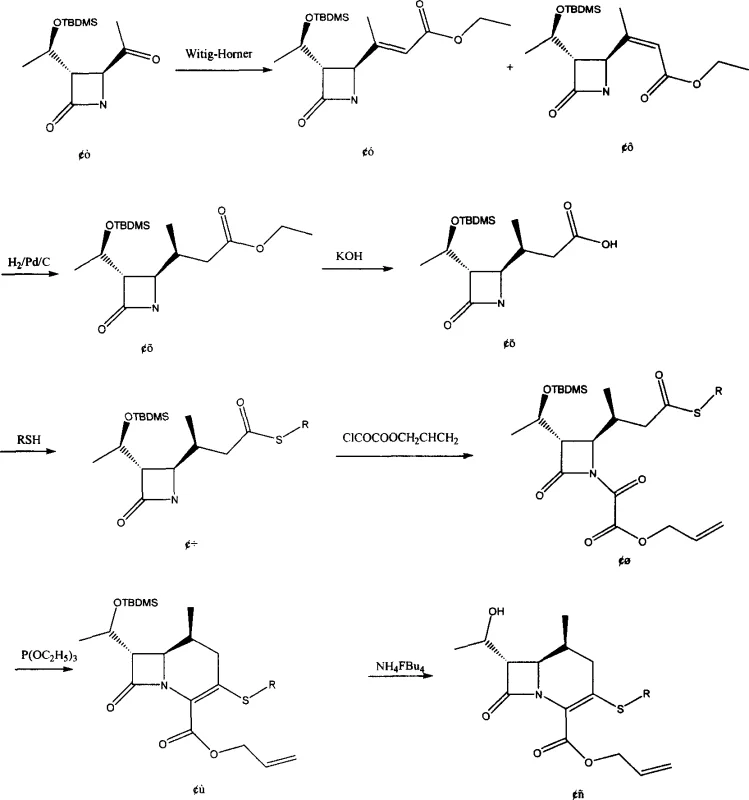
The mechanistic highlight of this patent is the transformation of the acyclic precursor (VIII) into the bicyclic carbacephem (IX). Upon treatment with triethyl phosphite, the molecule undergoes an intramolecular cyclization where the phosphite acts as a deoxygenating agent, facilitating the formation of the double bond within the six-membered ring while simultaneously closing the ring structure. This "carbonyl docking" mechanism avoids the generation of nitrogen gas and unstable intermediates associated with diazonium routes. The reaction is conducted in refluxing xylene with hydroquinone as a stabilizer, ensuring a clean conversion. Finally, the silyl protecting group is removed using tetrabutylammonium fluoride (TBAF) and acetic acid to reveal the free hydroxyl group, yielding the final target Compound (I). This sequence demonstrates exceptional control over regioselectivity and stereochemistry, which is paramount for a reliable agrochemical intermediate supplier or pharma partner aiming for consistent batch-to-batch quality.
How to Synthesize 1-Methyl Carbacephem Efficiently
Implementing this synthesis requires careful attention to reaction conditions, particularly during the olefination and cyclization stages. The initial Wittig-Horner step must be managed to optimize the ratio of trans to cis isomers, although the subsequent hydrogenation step converges these isomers into a single product, simplifying the purification requirements. The activation of the carboxylic acid to the thioester using CDI is a mild and efficient method that avoids the use of harsh chlorinating agents, preserving the integrity of the sensitive beta-lactam ring. For the critical cyclization step, maintaining anhydrous conditions and the correct stoichiometry of triethyl phosphite is essential to drive the reaction to completion without forming phosphorus-containing byproducts that are difficult to remove. The detailed standardized synthesis steps for this process are outlined in the guide below.
- Perform Wittig-Horner reaction on azetidinone with ethyl diethoxyphosphonoacetate using NaH to form olefin intermediates.
- Execute catalytic hydrogenation followed by alkaline hydrolysis to generate the carboxylic acid precursor.
- Form the thioester using CDI and thiophenol, then acylate with monoallyl oxalyl chloride.
- Conduct ring closure using triethyl phosphite and hydroquinone in refluxing xylene.
- Remove the silyl protecting group using tetrabutylammonium fluoride to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented route offers distinct advantages that directly address the concerns of procurement managers and supply chain heads regarding cost, safety, and scalability. By eliminating the need for hazardous diazonium salts, the process significantly lowers the barrier for entry for manufacturing facilities that may not have specialized explosion-proof infrastructure. This inherently safer design translates to reduced insurance premiums and lower capital expenditure requirements for plant modifications. Furthermore, the use of commodity chemicals such as triethyl phosphite, sodium hydride, and palladium on carbon ensures that raw material sourcing is stable and not subject to the volatility associated with specialized, low-volume reagents. This stability is crucial for reducing lead time for high-purity pharmaceutical intermediates and ensuring continuous supply for downstream API production.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts often required in older coupling methods, combined with the avoidance of expensive diazonium precursors, leads to a leaner cost structure. The convergence of cis and trans isomers during the hydrogenation step means that rigorous chromatographic separation after the first step is less critical, saving significant solvent and silica costs. Additionally, the ability to carry forward crude intermediates like Compound VIII without purification, due to their instability, streamlines the workflow and reduces unit operations, resulting in substantial cost savings in the overall production budget.
- Enhanced Supply Chain Reliability: The reliance on robust, well-understood chemical transformations such as hydrogenation and ester hydrolysis ensures that the process is less prone to unexpected failures or batch rejections. The reagents used, including potassium hydroxide and thiophenols, are widely available from multiple global suppliers, mitigating the risk of single-source bottlenecks. This diversification of the supply base enhances the resilience of the manufacturing chain, allowing for better planning and inventory management. Consequently, partners can expect more predictable delivery schedules and a lower risk of production stoppages due to raw material shortages.
- Scalability and Environmental Compliance: The process operates primarily at ambient pressure or under reflux, avoiding the need for high-pressure autoclaves or cryogenic reactors that limit batch sizes. This thermal profile is ideal for scaling from pilot plant to multi-ton commercial production. Moreover, the absence of heavy metal waste streams and explosive byproducts simplifies effluent treatment and regulatory compliance. The greener profile of this synthesis aligns with modern environmental, social, and governance (ESG) goals, making it a preferred choice for companies aiming to minimize their ecological footprint while maintaining high production volumes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the technical specifications and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of the method. Understanding these details is essential for technical teams evaluating the transfer of this technology to their own manufacturing sites or for assessing the quality of outsourced intermediates.
Q: What is the primary advantage of this synthesis route over conventional methods?
A: Unlike conventional routes relying on hazardous diazonium salts and L-aspartic acid, this method utilizes a safer triethyl phosphite-mediated cyclization, significantly reducing process risks and simplifying purification.
Q: Which reagents are critical for the ring-closing step?
A: The key cyclization step employs triethyl phosphite as the primary reagent along with a catalytic amount of hydroquinone in refluxing xylene to achieve carbonyl docking and ring formation.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the process avoids extreme cryogenic conditions and unstable intermediates, utilizing standard reagents like Pd/C and KOH, which supports robust commercial scale-up and supply chain reliability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Methyl Carbacephem Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving antibiotics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN101054376A can be translated into reliable industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify the stereochemical integrity and impurity profile of every batch. Our commitment to technical excellence ensures that the complex phosphite-mediated cyclization and preceding steps are executed with precision, delivering a product that meets the exacting standards of global regulatory bodies.
We invite you to collaborate with us to optimize your supply chain for carbacephem antibiotics. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage potential partners to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data and proven capability. Let us be your strategic partner in bringing efficient and safe antibiotic solutions to the market.