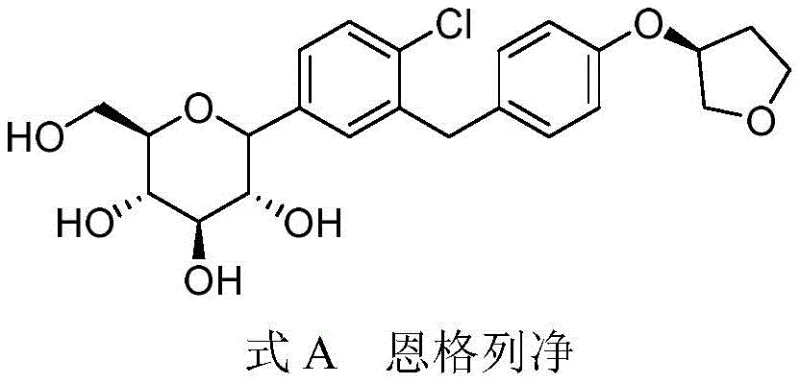
Advanced Synthesis of Empagliflozin Impurity I for Global Pharmaceutical Quality Control
The pharmaceutical industry's relentless pursuit of safety and efficacy in small molecule drug synthesis is exemplified by the rigorous control of impurities in high-value therapeutics like Empagliflozin. As a leading SGLT2 inhibitor, Empagliflozin requires meticulous quality assurance to ensure patient safety, necessitating the availability of certified reference standards for every potential degradation product or process-related impurity. Chinese patent CN114380774A introduces a groundbreaking synthesis method specifically designed to produce Empagliflozin Impurity I, a critical byproduct that can arise during the manufacturing of the active pharmaceutical ingredient. This innovation addresses a significant gap in the market, where previously, no literature reports existed for the specific preparation of this impurity, thereby complicating quality control protocols for generic and brand-name manufacturers alike. By establishing a reliable pathway to generate this reference standard, the patent empowers quality control laboratories to set accurate detection limits and ensure that the final drug product remains within safe therapeutic windows. The ability to synthesize this impurity on demand transforms it from an unknown risk into a quantifiable parameter, significantly enhancing the overall robustness of the drug's regulatory filing and commercial lifecycle management.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
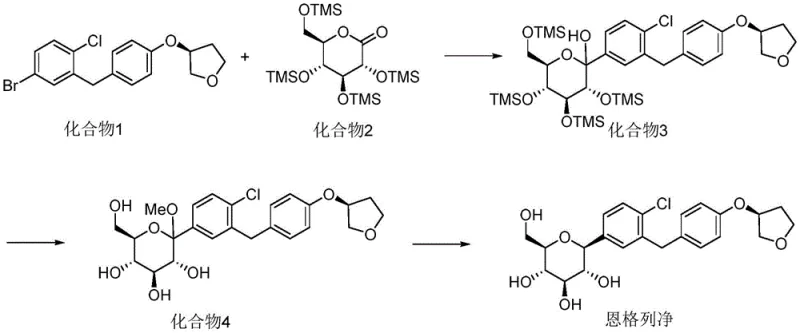
Historically, the synthesis of Empagliflozin has involved complex multi-step sequences that inadvertently generate trace impurities which are difficult to isolate and characterize without a dedicated synthesis route. Conventional approaches often rely on the primary production line to generate these impurities as minor byproducts, which are then painstakingly separated using preparative HPLC, a process that is notoriously inefficient, costly, and time-consuming for large-scale quality control needs. The lack of a specific synthetic method means that supply chains for reference standards are fragile, often relying on limited stockpiles that may degrade over time or vary in purity between batches. Furthermore, attempting to isolate these impurities from the main reaction mixture often exposes the material to harsh purification conditions that can alter its chemical structure, rendering it unsuitable as a stable reference standard. This uncertainty creates a bottleneck for pharmaceutical companies aiming to validate their analytical methods, as they cannot guarantee the identity or purity of the impurity standards used for calibration. Consequently, the absence of a dedicated synthesis route has long been a hidden vulnerability in the supply chain for SGLT2 inhibitor manufacturing, posing risks to regulatory compliance and batch release timelines.
The Novel Approach
The method disclosed in patent CN114380774A represents a paradigm shift by treating the impurity not as a waste product, but as a target molecule worthy of a dedicated, optimized synthetic pathway. Instead of relying on chance formation during the main drug synthesis, this novel approach utilizes a direct lithiation strategy starting from a specific brominated precursor, Compound 1, to construct the impurity structure with high precision. This targeted synthesis allows for the deliberate introduction of the butyl group at the specific position required to mimic the process-related impurity, ensuring that the resulting material is chemically identical to the trace contaminants found in production batches. By decoupling the impurity synthesis from the main drug manufacturing process, companies can produce large quantities of reference standards without interfering with API production schedules or compromising the yield of the active drug. This method also offers superior control over the stereochemistry and purity of the final product, as the reaction conditions are tailored specifically for this transformation rather than being a compromise within a broader synthetic scheme. Ultimately, this approach provides a stable, scalable, and reproducible source of Empagliflozin Impurity I, securing the quality control infrastructure for manufacturers worldwide.
Mechanistic Insights into Lithium-Halogen Exchange and Alkylation
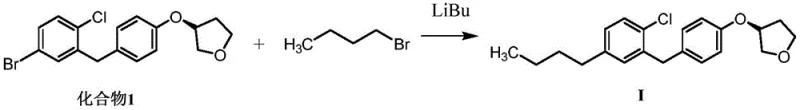
The core chemical transformation driving this synthesis is a low-temperature lithium-halogen exchange reaction, a powerful tool in organic synthesis that allows for the generation of highly reactive organolithium intermediates with excellent regioselectivity. In this specific protocol, Compound 1, which contains a bromine atom on the aromatic ring, is treated with n-butyllithium at cryogenic temperatures ranging from -70°C to -75°C. This extreme cold is not merely a precaution but a critical mechanistic requirement to stabilize the resulting aryl-lithium species and prevent unwanted side reactions such as nucleophilic attack on the sensitive ether linkages or the tetrahydrofuran ring present in the molecule. Once the lithium-halogen exchange is complete, the highly nucleophilic aryl-lithium intermediate is poised to react with an electrophile, in this case, bromobutane, which serves as the source of the butyl chain found in the impurity structure. The subsequent alkylation step proceeds smoothly as the carbon-lithium bond attacks the carbon-bromine bond of the butyl bromide, displacing the bromide ion and forming the new carbon-carbon bond that defines the impurity's structure. This mechanism ensures that the butyl group is installed exclusively at the position previously occupied by the bromine atom, guaranteeing the structural fidelity of the synthesized impurity relative to the one observed in drug substance batches.
Controlling impurities in such a complex molecule requires a deep understanding of how reaction conditions influence the formation of byproducts, and this patent leverages that understanding to maximize purity. The use of anhydrous tetrahydrofuran as the solvent is crucial, as any presence of moisture would instantly quench the reactive organolithium intermediate, leading to the formation of the de-halogenated starting material rather than the desired alkylated product. Furthermore, the stoichiometry is carefully balanced, with a slight excess of n-butyllithium (1.2 equivalents) ensuring complete conversion of the starting bromide, while the bromobutane is used in significant excess (2.2 equivalents) to drive the alkylation to completion. The quenching process using ammonium chloride solution is gentle yet effective, neutralizing any remaining organometallic species without causing hydrolysis of the acetal or ether functionalities that are sensitive to strong acids or bases. Following the reaction, purification via silica gel column chromatography using a specific eluent system of petroleum ether and ethyl acetate (50:1 ratio) allows for the separation of the target impurity from unreacted starting materials and minor side products. This rigorous attention to mechanistic detail results in a final product with an HPLC purity of 96.66%, making it highly suitable for use as a certified reference standard in analytical laboratories.
How to Synthesize Empagliflozin Impurity I Efficiently
The synthesis of Empagliflozin Impurity I is a precise operation that demands strict adherence to the patented protocol to ensure the generation of a high-quality reference standard suitable for regulatory submissions. The process begins with the preparation of a reaction vessel equipped for low-temperature chemistry, where Compound 1 is dissolved in anhydrous tetrahydrofuran and cooled to a precise range of -70°C to -75°C using a dry ice-acetone bath. This thermal control is the cornerstone of the reaction's success, as it governs the stability of the intermediate and the selectivity of the subsequent alkylation. Once the temperature is stabilized, n-butyllithium is added dropwise to initiate the lithium-halogen exchange, followed by the addition of bromobutane to complete the carbon-carbon bond formation. The reaction mixture is then allowed to warm to room temperature gradually, ensuring that the alkylation proceeds to completion before the mixture is quenched and worked up. For a comprehensive guide on the exact molar ratios, stirring times, and purification techniques required to replicate this synthesis in a GMP environment, please refer to the standardized protocol detailed below.
- Dissolve Compound 1 in anhydrous tetrahydrofuran and cool the mixture to -70°C to -75°C using a dry ice-acetone bath.
- Dropwise add n-butyllithium catalyst under inert atmosphere and maintain temperature for 1 hour to facilitate lithium-halogen exchange.
- Introduce bromobutane, stir at constant temperature, warm to room temperature, and purify via silica gel chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis method offers substantial strategic advantages that extend far beyond the laboratory bench, directly impacting the bottom line and operational resilience. By securing a dedicated source for Empagliflozin Impurity I, pharmaceutical companies can eliminate the dependency on unpredictable isolation from production batches, which often leads to supply shortages and delays in quality control testing. This reliability translates into a more streamlined regulatory approval process, as consistent access to high-purity reference standards ensures that analytical methods are validated without interruption, preventing costly batch holds or rejection. Furthermore, the simplicity of the synthetic route, which utilizes commercially available starting materials and standard reagents, implies a significantly reduced cost of goods sold for the reference standard itself compared to complex isolation procedures. This cost efficiency allows quality control budgets to be allocated more effectively, while the robust nature of the synthesis ensures that supply can be scaled rapidly to meet the demands of global clinical trials or commercial production without the need for specialized equipment.
- Cost Reduction in Manufacturing: The elimination of complex isolation steps and the use of readily available reagents like bromobutane and n-butyllithium drastically simplify the production process, leading to substantial cost savings in the procurement of reference standards. By avoiding the need for preparative HPLC purification from crude drug substance, manufacturers can reduce solvent consumption and labor hours, resulting in a more economical supply of critical quality control materials. This efficiency allows for the allocation of resources towards other critical areas of drug development, enhancing the overall financial viability of the SGLT2 inhibitor project.
- Enhanced Supply Chain Reliability: The ability to synthesize this impurity on demand ensures a continuous and reliable supply chain, mitigating the risks associated with single-source suppliers or limited stockpiles of isolated impurities. This independence from production batch variability means that quality control laboratories can maintain their testing schedules without interruption, regardless of the manufacturing status of the active pharmaceutical ingredient. Such reliability is crucial for maintaining compliance with strict regulatory timelines and ensuring that drug products reach the market without delay due to analytical bottlenecks.
- Scalability and Environmental Compliance: The synthetic route is designed for scalability, utilizing standard reaction conditions that can be easily transferred from laboratory to pilot plant scales without significant re-optimization. Additionally, the use of common solvents like tetrahydrofuran and ethyl acetate simplifies waste management and solvent recovery processes, aligning with modern environmental compliance standards and reducing the ecological footprint of reference standard production. This scalability ensures that as the demand for Empagliflozin grows, the supply of its critical impurity standards can grow in tandem, supporting long-term commercial success.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of Empagliflozin Impurity I, providing clarity for stakeholders involved in the quality control and manufacturing of SGLT2 inhibitors. These answers are derived directly from the technical specifications and beneficial effects outlined in the patent data, ensuring that the information provided is accurate and relevant to industry needs. Understanding these details is essential for making informed decisions about the integration of this synthesis method into your quality assurance workflows.
Q: Why is synthesizing Empagliflozin Impurity I critical for drug safety?
A: Synthesizing this specific impurity allows manufacturers to establish precise analytical limits, ensuring that the final SGLT2 inhibitor medication meets stringent global regulatory purity standards and avoids toxicological risks.
Q: What are the key reaction conditions for this synthesis?
A: The process requires strict low-temperature control between -70°C and -75°C using n-butyllithium in THF, followed by quenching with ammonium chloride to ensure high selectivity and yield.
Q: How does this method improve supply chain stability?
A: By utilizing readily available starting materials like Compound 1 and bromobutane, this route avoids complex multi-step sequences, reducing lead times and ensuring consistent availability of reference standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Empagliflozin Impurity Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your pharmaceutical products relies heavily on the quality and availability of your reference standards, which is why we have integrated this advanced synthesis technology into our CDMO capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demand for Empagliflozin Impurity I with the same rigor and precision as the active ingredient itself. We operate under stringent purity specifications and utilize rigorous QC labs to verify every batch, guaranteeing that the material you receive is fit for purpose in your analytical methods and regulatory filings. By partnering with us, you gain access to a supply chain that is not only robust and scalable but also deeply knowledgeable about the specific chemical challenges associated with SGLT2 inhibitor impurities.
We invite you to discuss how our technical expertise can optimize your quality control strategy and reduce your overall operational costs through a Customized Cost-Saving Analysis. Our technical procurement team is ready to provide you with specific COA data and route feasibility assessments tailored to your project's unique requirements, ensuring that you have the data you need to move forward with confidence. Contact us today to secure a reliable supply of high-purity Empagliflozin Impurity I and strengthen your position in the global pharmaceutical market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →