Scalable Production of High-Purity Antibacterial N-Formyl Hydroxylamine Intermediates via Novel Resolution and Deprotection
The pharmaceutical industry's relentless pursuit of novel antimicrobial agents has placed Peptide Deformylase (PDF) inhibitors at the forefront of antibacterial research, particularly for combating resistant bacterial strains. Patent CN1759097A discloses a groundbreaking chemical process for the preparation of intermediates essential for synthesizing N-[1-oxo-2-alkyl-3-(N-hydroxyformamido)-propyl] derivatives, which act as potent PDF inhibitors. This technology represents a significant leap forward in process chemistry by addressing critical bottlenecks such as safety hazards associated with oxidants and the challenging selectivity issues during final deprotection steps. By utilizing specific beta-lactam intermediates and enantiomerically pure resolving agents like (R)-2-butyl-3-hydroxy-propionic acid, the method ensures high optical purity essential for biological activity. Furthermore, the innovation avoids the use of unstable hydrogen peroxide and introduces a highly selective debenzylation protocol that minimizes unwanted by-products, thereby streamlining the path to commercial viability for these complex antibacterial intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of N-formyl hydroxylamine compounds relied on oxidative conditions that introduced significant safety and purity challenges for large-scale manufacturing. Conventional routes often necessitated the use of hydrogen peroxide (H2O2) for specific oxidation or deprotection steps, a reagent known for its thermal instability and potential for runaway reactions, especially when handling large batches in industrial reactors. Moreover, the final deprotection stages typically involved catalytic hydrogenation under standard atmospheric or elevated hydrogen pressures. A critical flaw in these traditional methods was the lack of chemoselectivity; the vigorous hydrogenation conditions frequently caused the unintended hydrogenolysis of the sensitive N-O bond within the pyridine N-oxide moiety. This side reaction generated substantial quantities of a difficult-to-separate impurity known as 'deoxy C10', which compromised the overall yield and required extensive, costly purification efforts such as preparative HPLC or multiple recrystallizations to meet pharmaceutical grade specifications.
The Novel Approach
The process detailed in CN1759097A fundamentally reengineers the synthetic pathway to overcome these legacy limitations through strategic chemical design and condition optimization. A cornerstone of this novel approach is the utilization of a specific chiral resolving agent, (R)-2-butyl-3-hydroxy-propionic acid, which is prepared via a robust resolution process using (R)-alpha-methylbenzylamine. 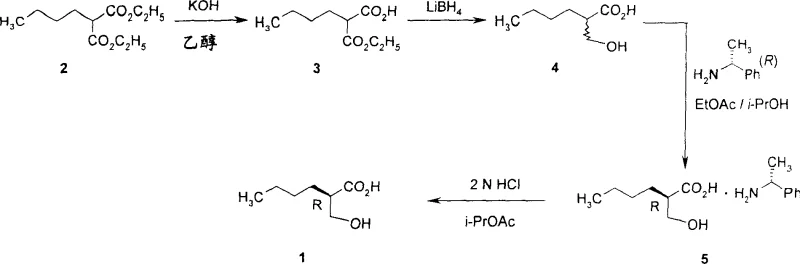 This resolution strategy ensures the establishment of the correct stereochemistry early in the synthesis, which is critical for the biological efficacy of the final PDF inhibitor. Furthermore, the process completely eliminates the need for hydrogen peroxide, replacing it with safer alternative reagents and conditions. Perhaps most notably, the invention introduces a selective debenzylation technique that operates either under sub-atmospheric hydrogen partial pressures or via transfer hydrogenation using formic acid and 4-methylmorpholine. This gentle deprotection method preserves the integrity of the N-O bond, effectively suppressing the formation of the 'deoxy C10' impurity and resulting in a much cleaner crude product profile that is amenable to simpler isolation techniques.
This resolution strategy ensures the establishment of the correct stereochemistry early in the synthesis, which is critical for the biological efficacy of the final PDF inhibitor. Furthermore, the process completely eliminates the need for hydrogen peroxide, replacing it with safer alternative reagents and conditions. Perhaps most notably, the invention introduces a selective debenzylation technique that operates either under sub-atmospheric hydrogen partial pressures or via transfer hydrogenation using formic acid and 4-methylmorpholine. This gentle deprotection method preserves the integrity of the N-O bond, effectively suppressing the formation of the 'deoxy C10' impurity and resulting in a much cleaner crude product profile that is amenable to simpler isolation techniques.
Mechanistic Insights into Asymmetric Hydrogenation and Selective Deprotection
The stereochemical integrity of the antibacterial intermediate is established through a sophisticated asymmetric hydrogenation step, which serves as a pivotal moment in the synthetic sequence. This transformation involves the reduction of an olefinic precursor, such as a methyl 2-[[(phenylmethoxy)amino]methyl]-2-hexenoate derivative, using a homogeneous rhodium catalyst system. The catalyst typically comprises a rhodium source, such as bis(norbornadiene)rhodium(I) tetrafluoroborate, coordinated with a chiral diphosphine ligand like (1R,1'R,2S,2'S)-TangPhos or (2S,5S)-Me-Duphos. The mechanism proceeds through the coordination of the olefin substrate to the chiral metal center, followed by oxidative addition of hydrogen and migratory insertion. The steric bulk and electronic properties of the chiral ligand create a highly differentiated environment around the metal, directing the hydride delivery to one specific face of the double bond. This results in the formation of the desired (S)-enantiomer with exceptional enantiomeric excess (often >98% ee), ensuring that the downstream biological activity is maximized while minimizing the presence of the inactive or potentially toxic antipode.
The final stage of the synthesis involves the removal of the benzyl protecting group to reveal the active hydroxamic acid functionality, a step that requires exquisite chemoselectivity to avoid damaging the molecule's pharmacophore. 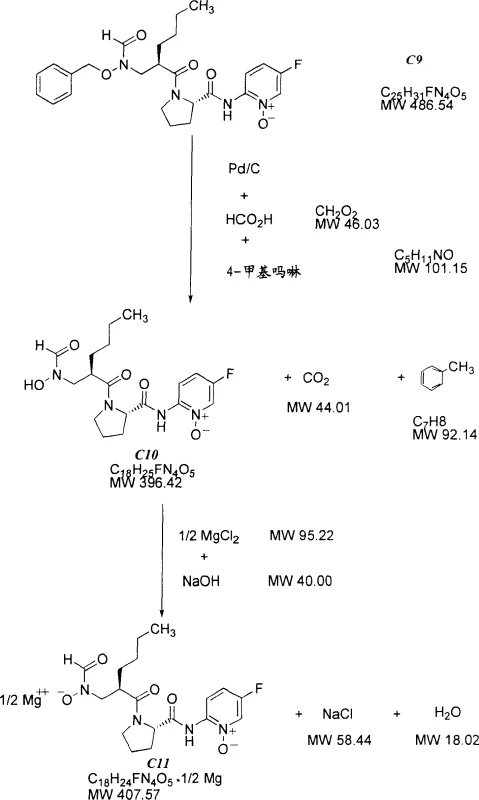 The patent elucidates a mechanism where the reaction kinetics are tuned to favor O-benzyl cleavage over N-O bond reduction. In the transfer hydrogenation variant, formic acid serves as the hydrogen donor in the presence of a palladium catalyst and a base like 4-methylmorpholine. The formic acid decomposes on the catalyst surface to generate reactive hydride species in situ, which selectively attack the benzylic carbon-oxygen bond. Alternatively, when using molecular hydrogen, the process strictly controls the hydrogen partial pressure to below 1 atmosphere (e.g., 0.1 to 0.24 atm) by diluting with nitrogen. This kinetic control ensures that the concentration of dissolved hydrogen is sufficient to reduce the benzyl ether but too low to drive the thermodynamically more demanding reduction of the pyridine N-oxide, thereby achieving high selectivity and minimizing the generation of deoxygenated by-products.
The patent elucidates a mechanism where the reaction kinetics are tuned to favor O-benzyl cleavage over N-O bond reduction. In the transfer hydrogenation variant, formic acid serves as the hydrogen donor in the presence of a palladium catalyst and a base like 4-methylmorpholine. The formic acid decomposes on the catalyst surface to generate reactive hydride species in situ, which selectively attack the benzylic carbon-oxygen bond. Alternatively, when using molecular hydrogen, the process strictly controls the hydrogen partial pressure to below 1 atmosphere (e.g., 0.1 to 0.24 atm) by diluting with nitrogen. This kinetic control ensures that the concentration of dissolved hydrogen is sufficient to reduce the benzyl ether but too low to drive the thermodynamically more demanding reduction of the pyridine N-oxide, thereby achieving high selectivity and minimizing the generation of deoxygenated by-products.
How to Synthesize N-Formyl Hydroxylamine Intermediates Efficiently
The implementation of this advanced synthetic route requires precise control over reaction parameters to maximize yield and optical purity while maintaining operational safety. The process begins with the preparation of the chiral acid resolving agent, followed by the coupling and cyclization steps to form the core beta-lactam or acyclic scaffold. Subsequent functionalization involves the installation of the hydroxylamine moiety and the final formylation. Each step is optimized for solvent compatibility and temperature control to prevent epimerization or degradation. The detailed standardized synthesis steps see the guide below for specific operational parameters regarding reagent stoichiometry, addition rates, and workup procedures.
- Prepare the chiral resolving agent (R)-2-butyl-3-hydroxypropionic acid via reduction and optical resolution using (R)-alpha-methylbenzylamine.
- Perform asymmetric hydrogenation of the olefinic intermediate using a Rhodium catalyst with chiral ligands like TangPhos to establish stereochemistry.
- Execute selective debenzylation using transfer hydrogenation with formic acid and 4-methylmorpholine to avoid N-O bond cleavage.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of the process described in CN1759097A offers tangible strategic advantages that extend beyond mere technical feasibility. The elimination of hazardous reagents like hydrogen peroxide simplifies the safety infrastructure required for production, potentially lowering insurance premiums and reducing the need for specialized containment equipment. This translates directly into cost reduction in pharmaceutical intermediate manufacturing by streamlining the regulatory compliance burden and minimizing the risk of production stoppages due to safety incidents. Furthermore, the enhanced selectivity of the debenzylation step significantly reduces the load on downstream purification units. By minimizing the formation of the 'deoxy C10' impurity, the process decreases the consumption of solvents and stationary phases required for chromatography or the energy costs associated with multiple recrystallizations, leading to a more sustainable and economically efficient production cycle.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by removing the need for expensive and hazardous oxidants, thereby reducing waste disposal costs and safety mitigation expenses. The high selectivity of the final deprotection step minimizes product loss during purification, effectively increasing the overall mass balance and yield of the active pharmaceutical ingredient. Additionally, the use of readily available starting materials and standard catalytic systems avoids reliance on exotic or supply-constrained reagents, stabilizing the raw material cost structure against market volatility.
- Enhanced Supply Chain Reliability: By utilizing robust chiral resolution techniques rather than relying solely on expensive chiral pool starting materials or complex enzymatic processes, the supply chain becomes more resilient and less susceptible to single-source bottlenecks. The ability to produce the key chiral intermediate in-house from commodity chemicals ensures a continuous and reliable flow of materials, reducing lead time for high-purity antibacterial intermediates. The scalability of the resolution and hydrogenation steps means that production volumes can be ramped up quickly to meet sudden surges in demand without compromising quality or delivery schedules.
- Scalability and Environmental Compliance: The transition to transfer hydrogenation or low-pressure hydrogenation for the final step significantly improves the environmental footprint of the manufacturing process. This approach reduces the emission of volatile organic compounds and lowers the energy consumption associated with high-pressure reactor operations. The process is inherently designed for commercial scale-up of complex pharmaceutical intermediates, with reaction conditions that are easily manageable in standard glass-lined or stainless steel reactors, facilitating a smooth technology transfer from pilot plant to full-scale commercial production facilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis route. These insights are derived directly from the experimental data and process descriptions within the patent documentation, providing clarity on safety, purity, and scalability concerns that are critical for decision-makers evaluating this technology for their supply chains.
Q: How does the new process improve safety compared to prior art methods?
A: The novel process eliminates the use of hydrogen peroxide (H2O2), which is unstable and poses safety risks during scale-up. Additionally, it employs transfer hydrogenation or low-pressure hydrogenation for the final deprotection step, significantly reducing the risk of explosive gas mixtures associated with high-pressure hydrogenation.
Q: What is the advantage of the selective debenzylation method described?
A: Traditional hydrogenation often cleaves the sensitive N-O bond in the pyridine N-oxide moiety, creating difficult-to-remove 'deoxy C10' impurities. The patented method uses controlled hydrogen partial pressure (<1 atm) or formic acid transfer hydrogenation to selectively remove the benzyl group while preserving the N-O bond, drastically improving purity.
Q: Is the chiral resolution process scalable for commercial production?
A: Yes, the process utilizes classical diastereomeric salt formation with (R)-alpha-methylbenzylamine followed by recrystallization. This is a robust, well-understood unit operation in the fine chemical industry that can be easily scaled from kilograms to multi-ton quantities without requiring expensive chiral chromatography.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable N-Formyl Hydroxylamine Intermediates Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust and scalable synthetic routes in the development of next-generation antibacterial agents. Our team of expert process chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN1759097A can be translated into reliable manufacturing reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of N-formyl hydroxylamine intermediates meets the highest standards of quality and consistency required by global regulatory bodies.
We invite you to collaborate with us to leverage this advanced technology for your antibiotic development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this optimized process can enhance your project's economics. Please contact us today to request specific COA data and route feasibility assessments, and let us partner with you to accelerate the delivery of life-saving antimicrobial therapies to the market.