Advanced Synthesis of 6-Aryloxymethyl-4-Aryl-3-Morpholinone Derivatives for Pharma
Introduction to Novel Morpholinone Derivative Technology
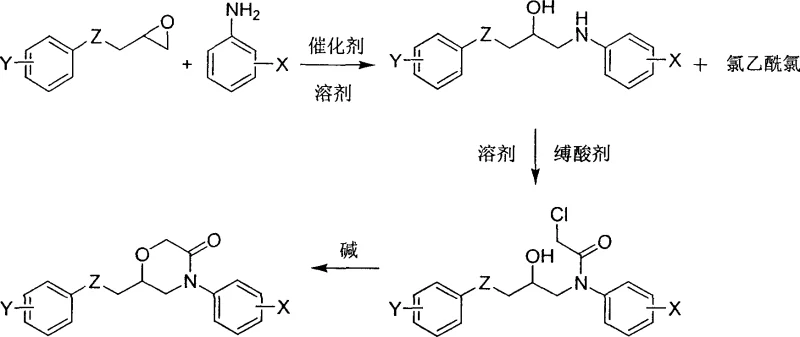
The pharmaceutical industry continuously seeks robust synthetic routes for heterocyclic compounds that serve as critical scaffolds for drug discovery. Patent CN1736992A introduces a significant advancement in the preparation of 6-aryloxymethyl-4-aryl-3-morpholinone derivatives, a class of compounds previously underexplored compared to their 2-morpholinone counterparts. While existing literature, such as US20030212062A1, focuses heavily on 2-morpholinone structures for lipid-lowering applications, this technology addresses the scarcity of efficient methods for synthesizing the 3-morpholinone isomer. The disclosed method offers a streamlined three-step pathway that bypasses the complex and hazardous multi-step sequences found in older patents like US6265402B1. By leveraging a heterogeneous alumina catalyst for the initial ring-opening reaction, the process achieves high selectivity and ease of purification, making it an attractive candidate for reliable pharmaceutical intermediate supplier networks seeking to diversify their portfolio with bioactive heterocycles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 3-morpholinone derivatives has been plagued by inefficient and dangerous chemical pathways. For instance, the methodology outlined in U.S. Pat 6265402B1 relies on a five-step sequence starting from ketones and trimethylsilyl cyanide, necessitating the handling of highly toxic hydrogen cyanide (HCN) equivalents. This not only imposes severe safety protocols and specialized equipment requirements but also generates substantial hazardous waste, complicating cost reduction in API manufacturing. Furthermore, alternative routes described in U.S. Pat 3308121, while avoiding cyanide, often suffer from limited substrate scope and rigorous reaction conditions that hinder scalability. The reliance on homogeneous catalysts or harsh reagents in these conventional methods frequently leads to difficult downstream processing, where removing trace metal impurities or byproducts requires extensive chromatographic purification, thereby increasing production lead times and operational expenditures for supply chain managers.
The Novel Approach
In stark contrast, the technology defined in CN1736992A presents a drastically simplified three-step protocol that enhances both safety and efficiency. The process initiates with the reaction of 3-aryloxy-1,2-epoxypropane and an arylamine compound using activated alumina as a catalyst. This heterogeneous catalytic system operates effectively under reflux conditions in common solvents like tetrahydrofuran or toluene, allowing for simple filtration to remove the catalyst post-reaction. The subsequent acylation with chloroacetyl chloride and final cyclization using sodium hydride proceed under controlled low-temperature conditions, ensuring high regioselectivity. This approach eliminates the need for toxic cyanide reagents entirely and reduces the total number of unit operations. For procurement teams, this translates to a more stable supply chain with reduced dependency on controlled substances, while the simplified workup procedures facilitate the commercial scale-up of complex pharmaceutical intermediates without compromising on purity standards.
Mechanistic Insights into Alumina-Catalyzed Cyclization
The core innovation of this synthesis lies in the mechanistic efficiency of the alumina-catalyzed ring-opening and the subsequent intramolecular cyclization. In the first step, the activated alumina surface acts as a Lewis acid-base pair, activating the epoxide ring of the 3-aryloxy-1,2-epoxypropane towards nucleophilic attack by the arylamine. This heterogeneous interaction promotes the formation of the beta-amino alcohol derivative with high fidelity, minimizing side reactions such as polymerization of the epoxide. Following acylation to form the 2-chloro-N-(2-hydroxyl-3-aryloxypropyl)-N-aryl acetamide, the final ring-closing step is critical. Under inert atmosphere and low temperatures ranging from -25°C to 0°C, a strong base like sodium hydride deprotonates the secondary hydroxyl group. The resulting alkoxide ion performs an intramolecular SN2 attack on the adjacent chloroacetyl carbon, displacing the chloride ion and closing the six-membered morpholinone ring. This precise control over reaction kinetics prevents intermolecular polymerization and ensures the formation of the desired 3-morpholinone core rather than alternative oligomers.
Furthermore, the impurity profile of this process is inherently cleaner due to the specificity of the cyclization step. The use of stoichiometric base ratios, typically between 1:1.1 to 1:2 relative to the substrate, ensures complete conversion while minimizing excess reagent carryover. The patent data indicates that purification can be achieved via standard silica gel chromatography using petroleum ether and ethyl acetate gradients, although for industrial applications, crystallization protocols can be developed based on the solid-state properties of the final derivatives, such as the light yellow solids observed in Embodiment 1 and 4. This mechanistic clarity provides R&D directors with confidence in the reproducibility of the route, as the reaction parameters—such as the 1 to 30 hour reflux time for the first step and the strict temperature control during cyclization—are well-defined and robust against minor fluctuations in raw material quality.
How to Synthesize 6-Aryloxymethyl-4-Aryl-3-Morpholinone Efficiently
To implement this synthesis effectively, operators must adhere to the specific stoichiometric and thermal conditions outlined in the patent embodiments. The process begins with the careful mixing of the epoxide and amine precursors in a polar or non-polar solvent, followed by the addition of the alumina catalyst. After the initial coupling, the intermediate beta-amino alcohol is isolated and immediately subjected to acylation under ice bath conditions to prevent thermal degradation. The final cyclization requires rigorous exclusion of moisture and oxygen, utilizing nitrogen protection and pre-cooled solvents to maintain the reaction temperature between -25°C and 0°C. Detailed standardized synthetic steps see the guide below.
- React 3-aryloxy-1,2-epoxypropane with an arylamine compound in the presence of activated alumina catalyst at reflux temperature to form a beta-amino alcohol derivative.
- Acylate the resulting beta-amino alcohol with chloroacetyl chloride using an acid binding agent like triethylamine or potassium carbonate under ice bath conditions.
- Perform intramolecular cyclization of the chloroacetamide intermediate using a strong base such as sodium hydride at low temperatures (-25°C to 0°C) to close the morpholinone ring.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this synthetic route offers substantial strategic benefits for organizations focused on cost reduction in pharmaceutical intermediate manufacturing. The elimination of hazardous cyanide reagents removes the need for expensive scrubbing systems and specialized waste disposal contracts, leading to significant overhead savings. Additionally, the use of activated alumina, a widely available and inexpensive heterogeneous catalyst, replaces costly transition metal catalysts that often require complex removal steps to meet residual metal specifications in active pharmaceutical ingredients. This simplification of the catalyst system directly contributes to a more streamlined production workflow, reducing the overall cycle time from raw material intake to finished goods. For supply chain heads, the reliance on commodity chemicals like chloroacetyl chloride, anilines, and epoxides ensures a stable and diversified sourcing strategy, mitigating the risk of supply disruptions associated with specialty reagents.
- Cost Reduction in Manufacturing: The process design inherently lowers operational expenditures by utilizing a heterogeneous catalyst that can be easily filtered off, eliminating the need for expensive aqueous quenching and extraction steps typically required for homogeneous catalysts. Furthermore, the avoidance of toxic HCN precursors reduces the regulatory compliance costs and insurance premiums associated with handling high-risk chemicals. The high yields reported in the embodiments, such as the 78% yield in the final cyclization step of Embodiment 1, indicate a material-efficient process that minimizes raw material waste. By reducing the number of synthetic steps from five in prior art to just three, the cumulative yield loss is significantly mitigated, resulting in a higher overall throughput per batch and better asset utilization for manufacturing facilities.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, including various substituted anilines and aryloxy-epoxides, are commercially available in bulk quantities from multiple global vendors. This commoditization of feedstocks prevents single-source bottlenecks and allows procurement managers to negotiate favorable pricing through competitive bidding. The robustness of the reaction conditions, which tolerate a range of solvents including methanol, ethanol, and toluene, provides flexibility in solvent sourcing, further insulating the supply chain from regional availability issues. Moreover, the stability of the intermediates allows for potential telescoping of steps or storage of key intermediates, providing buffer capacity against demand fluctuations and ensuring consistent delivery schedules to downstream API manufacturers.
- Scalability and Environmental Compliance: From an environmental perspective, this route aligns with green chemistry principles by avoiding persistent organic pollutants and heavy metals. The solid waste generated, primarily spent alumina, is non-hazardous and easier to dispose of compared to heavy metal sludge. The process is highly amenable to scale-up, as demonstrated by the clear parameter ranges for temperature and molar ratios provided in the patent. The exothermic nature of the acylation and cyclization steps is manageable with standard industrial cooling systems, reducing the risk of thermal runaway incidents. This safety profile facilitates faster regulatory approval for new manufacturing sites and simplifies the technology transfer process between R&D pilot plants and commercial production units, ensuring a smoother path to market for new drug candidates utilizing this scaffold.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these details is crucial for technical teams evaluating the feasibility of integrating this route into existing production lines or for sourcing teams assessing the quality capabilities of potential partners.
Q: What are the primary safety advantages of this synthesis route compared to prior art?
A: Unlike previous methods described in patents like US6265402B1 which utilize toxic trimethylsilyl cyanide and hydrogen cyanide (HCN) over five steps, this novel process avoids hazardous cyanide reagents entirely, significantly improving operational safety and reducing environmental compliance burdens.
Q: What specific catalyst is utilized in the initial ring-opening step?
A: The process employs activated alumina as a heterogeneous catalyst for the reaction between 3-aryloxy-1,2-epoxypropane and the arylamine. This allows for easy filtration and removal of the catalyst, simplifying the downstream purification process compared to homogeneous catalytic systems.
Q: What are the potential therapeutic applications of these derivatives?
A: According to the patent data, these 6-aryloxymethyl-4-aryl-3-morpholinone derivatives exhibit pharmacological activities including lipid-lowering effects, anti-inflammatory properties, and potential utility as cell death inducers or antitumor agents in chemical genetics research.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 6-Aryloxymethyl-4-Aryl-3-Morpholinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality heterocyclic intermediates play in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of 6-aryloxymethyl-4-aryl-3-morpholinone meets the exacting standards required for pharmaceutical applications. Our infrastructure is designed to handle the specific safety requirements of this chemistry, including the safe handling of sodium hydride and chloroacetyl chloride, providing a secure environment for your project.
We invite you to collaborate with us to leverage this advanced synthetic technology for your drug discovery programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and purity needs. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise in alumina-catalyzed synthesis can accelerate your timeline to market while optimizing your overall production costs.