Advanced Synthesis of Neuroprotective 3-Piperidino-1-Chromanol Derivatives for Commercial Pharmaceutical Production
The pharmaceutical landscape for central nervous system (CNS) disorders is constantly evolving, driven by the need for agents that offer therapeutic efficacy without debilitating side effects. Patent CN1053923A introduces a groundbreaking class of 3-piperidino-1-chroman alcohol derivatives that exhibit potent neuroprotective properties, specifically anti-ischemic and excitatory amino acid receptor blocking effects. Unlike earlier compounds such as Benzphenidol, which were primarily marketed as antihypertensives, these novel derivatives are engineered to minimize hypotensive activity while maximizing neuroprotection against conditions like stroke, Alzheimer's disease, and Parkinson's disease. This technological leap represents a significant opportunity for pharmaceutical manufacturers seeking to develop next-generation CNS therapeutics with improved safety profiles. The structural innovation lies in the specific substitution patterns on the chroman ring and the piperidine moiety, which decouple the desired neurological activity from cardiovascular side effects. As a reliable pharmaceutical intermediates supplier, understanding the nuances of this patent is crucial for integrating these high-value scaffolds into modern drug discovery pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the development of neuroprotective agents was hindered by the pleiotropic effects of early lead compounds, which often caused severe hypotension limiting their therapeutic window. Conventional synthesis routes for related phenolic piperidine derivatives frequently relied on harsh conditions that compromised stereochemical integrity, leading to complex mixtures of isomers that were difficult and costly to separate. The lack of selectivity in traditional methods meant that significant resources were expended on purification processes to remove impurities that could exacerbate cardiovascular risks in patients. Furthermore, older methodologies often utilized expensive transition metal catalysts or reagents that posed environmental hazards and supply chain vulnerabilities, driving up the cost reduction in pharmaceutical intermediates manufacturing. The inability to precisely control the substitution at the 3-position of the chroman ring resulted in batch-to-batch variability, complicating regulatory approval processes for high-purity pharmaceutical intermediates. These legacy challenges underscore the need for a more refined, selective, and scalable synthetic strategy that aligns with modern Good Manufacturing Practice (GMP) standards.
The Novel Approach
The methodology outlined in CN1053923A overcomes these historical barriers by employing a sophisticated sequence of nucleophilic substitution and selective hydride reduction. This novel approach utilizes protected intermediates, such as silyl ethers, to direct reactivity and prevent unwanted side reactions at phenolic hydroxyl groups during the critical bond-forming steps. By carefully selecting reaction conditions, such as temperature and solvent polarity, the process favors the formation of the desired cis or trans stereochemistry, significantly simplifying downstream purification. The use of readily available starting materials, including substituted chromanones and piperidine derivatives, ensures that the commercial scale-up of complex pharmaceutical intermediates is both economically viable and logistically robust. This strategy not only enhances the overall yield of the active pharmaceutical ingredient but also reduces the generation of hazardous waste, aligning with green chemistry principles. Consequently, this method provides a clear pathway for producing high-quality intermediates that meet the stringent requirements of global regulatory bodies.
Mechanistic Insights into Nucleophilic Substitution and Hydride Reduction
The core of this synthetic innovation revolves around the precise construction of the 3-piperidino-1-chroman scaffold, as defined by Formula (I). The reaction mechanism typically begins with the activation of a chromanone precursor, often via alpha-bromination, to create an electrophilic center susceptible to nucleophilic attack by a substituted piperidine. This step is critical for establishing the carbon-nitrogen bond that links the heterocyclic rings, and the use of a tertiary amine base facilitates the displacement of the halide leaving group without promoting elimination side reactions. The stereochemical outcome is influenced by the steric bulk of the substituents on both the chroman ring and the piperidine nitrogen, allowing for the preferential formation of either cis or trans isomers depending on the specific therapeutic requirement. Understanding these mechanistic details is essential for optimizing reaction parameters to achieve the highest possible purity and yield.
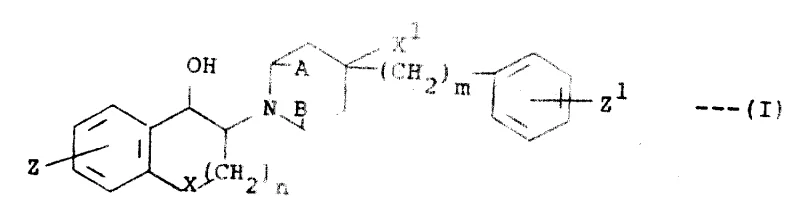
Following the formation of the ketone intermediate, the process proceeds to a stereoselective reduction step using hydride reagents such as sodium borohydride or lithium aluminum hydride. This reduction converts the ketone functionality into the corresponding alcohol, establishing the second chiral center at the 1-position of the chroman ring. The choice of reducing agent and reaction temperature plays a pivotal role in determining the diastereomeric ratio, with milder conditions often favoring the kinetic product while thermodynamic control can be achieved through equilibration. Subsequent removal of protecting groups, such as triisopropylsilyl ethers, is accomplished under mild fluoride conditions to reveal the free phenolic hydroxyl without affecting the sensitive amine or alcohol functionalities. This multi-step sequence ensures that the final product retains the structural integrity required for potent neuroprotective activity while minimizing the presence of genotoxic impurities.
How to Synthesize 3-Piperidino-1-Chromanol Derivatives Efficiently
Implementing this synthesis on an industrial scale requires a thorough understanding of the critical process parameters identified in the patent literature. The route generally involves the preparation of a protected alpha-amino ketone intermediate, followed by nucleophilic substitution with the appropriate piperidine derivative and final reduction. Each step must be carefully monitored to ensure that reaction completion is achieved without degrading the sensitive functional groups present in the molecule. Detailed standardized synthesis steps are provided below to guide process chemists in replicating these results with high fidelity and consistency.
- Prepare the protected alpha-amino ketone intermediate using silyl protecting groups to ensure regioselectivity.
- Execute nucleophilic substitution with substituted piperidines in the presence of a tertiary amine base.
- Perform hydride reduction followed by deprotection to yield the final high-purity chromanol diol.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the synthetic route described in CN1053923A offers substantial benefits for procurement and supply chain management teams looking to optimize their sourcing strategies. The reliance on commodity chemicals and standard organic transformations means that the supply chain is less susceptible to disruptions caused by the scarcity of exotic reagents or specialized catalysts. This robustness translates into greater supply continuity and reduced risk of production delays, which are critical factors for maintaining the timelines of drug development programs. Furthermore, the elimination of complex purification steps associated with isomer separation significantly lowers the overall manufacturing cost, making these intermediates more accessible for large-scale production. By adopting this efficient synthesis, companies can achieve significant cost savings while ensuring a steady supply of high-quality materials for their clinical and commercial needs.
- Cost Reduction in Manufacturing: The synthetic pathway avoids the use of expensive transition metal catalysts and precious metal scavengers, which are often major cost drivers in pharmaceutical manufacturing. Instead, it relies on cost-effective reagents like sodium borohydride and common organic bases, which drastically simplifies the bill of materials. The high yields reported in the examples, such as the 84% yield in the deprotection step, indicate a material-efficient process that minimizes waste disposal costs. Additionally, the ability to perform reactions at ambient or slightly elevated temperatures reduces energy consumption associated with heating and cooling, further contributing to overall operational expenditure reductions. These factors combine to create a highly economical process that enhances the profit margin for the final drug product.
- Enhanced Supply Chain Reliability: The starting materials required for this synthesis, including substituted chromanones and piperidines, are widely available from multiple global suppliers, reducing dependency on single-source vendors. This diversification of the supply base mitigates the risk of shortages and allows for more flexible procurement negotiations. The robustness of the chemical steps means that the process is tolerant to minor variations in raw material quality, ensuring consistent output even when sourcing from different batches or suppliers. Consequently, this reliability supports reducing lead time for high-purity pharmaceutical intermediates, enabling faster progression from preclinical studies to clinical trials. A stable supply chain is essential for meeting the demanding schedules of modern drug development.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing solvents and conditions that are easily managed in large-scale reactors without significant safety hazards. The absence of highly toxic reagents simplifies waste treatment and disposal, ensuring compliance with increasingly stringent environmental regulations. The use of standard workup procedures, such as aqueous washes and crystallization, facilitates easy technology transfer from the laboratory to pilot and commercial plants. This scalability ensures that the manufacturing capacity can be rapidly expanded to meet market demand without the need for specialized equipment or infrastructure investments. Such environmental and operational efficiency is a key advantage for sustainable pharmaceutical manufacturing.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these neuroprotective intermediates. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation to ensure accuracy and relevance. Understanding these details helps stakeholders make informed decisions about integrating this technology into their product portfolios.
Q: How do these derivatives differ from Benzphenidol in terms of side effects?
A: Unlike Benzphenidol, which exhibits significant hypotensive effects, the compounds of Formula (I) provide selective neuroprotection with little to no blood pressure reduction, making them safer for CNS indications.
Q: What is the stereochemical preference in the synthesis of these intermediates?
A: The synthesis typically yields a mixture of cis and trans isomers, with the cis isomer often predominating. Separation can be achieved via chromatography or fractional crystallization.
Q: Are the starting materials for this synthesis commercially scalable?
A: Yes, the starting materials such as substituted chromanones and piperidine derivatives are readily available or easily synthesized from commodity chemicals, supporting large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Piperidino-1-Chromanol Derivatives Supplier
At NINGBO INNO PHARMCHEM, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop to market. Our team of expert chemists is well-versed in the nuances of CNS active pharmaceutical ingredients and can navigate the complexities of stereochemical control and impurity profiling inherent in this synthesis. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch meets the highest international standards for safety and efficacy. Our commitment to quality ensures that you receive materials that are ready for immediate use in formulation or further synthetic elaboration without additional purification burdens.
We invite you to contact our technical procurement team to discuss your specific requirements and explore how we can support your supply chain goals. By requesting a Customized Cost-Saving Analysis, you can gain valuable insights into how our manufacturing capabilities can optimize your budget and timeline. We are prepared to provide specific COA data and route feasibility assessments to demonstrate our capability to deliver on your project milestones. Partnering with us means gaining a strategic ally dedicated to the success of your neuroprotective drug development program.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →