Advanced Manufacturing of High-Purity Ondansetron Hydrochloride Dihydrate for Global Pharma Supply Chains
Introduction to Advanced Ondansetron Manufacturing Technologies
The pharmaceutical landscape demands increasingly rigorous standards for Active Pharmaceutical Ingredients (APIs), particularly for critical antiemetic agents like ondansetron used in oncology support care. Patent CN1496350A discloses a transformative methodology for the preparation of pure ondansetron hydrochloride dihydrate, addressing long-standing challenges regarding impurity profiles and chemical homogeneity. This intellectual property outlines a robust synthetic route that begins with the efficient generation of dimethylaminomethylcarbazolone and culminates in a novel recrystallization technique capable of delivering product purity exceeding 99.0%. For R&D directors and procurement specialists, understanding the nuances of this process is vital, as it represents a significant leap forward in cost reduction in API manufacturing while ensuring the highest levels of patient safety through superior impurity control. The technology specifically targets the elimination of the notorious exo-methylene by-product, a persistent contaminant in earlier generations of synthesis that compromised both the stability and the regulatory compliance of the final drug substance.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of ondansetron has been plagued by the formation of structurally related impurities that are difficult to separate using standard purification techniques. Prior art, such as the methods described in US Patent 4,695,578, often resulted in final products containing detectable levels of 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one, commonly referred to as the exo-methylene by-product. This impurity not only affects the color and aesthetic quality of the API but also raises concerns regarding the overall safety profile and shelf-life of the medication. Conventional processes frequently relied on complex solvent systems and multiple recrystallization steps that drove up production costs and extended lead times, creating bottlenecks for commercial scale-up of complex pharmaceutical intermediates. Furthermore, the reliance on organic solvents for the final salt formation often introduced additional risks of solvent residues, necessitating extensive drying and testing protocols that further strained manufacturing resources and delayed time-to-market for generic formulations.
The Novel Approach
The improved process detailed in the patent data introduces a streamlined workflow that fundamentally alters the purification landscape for this critical molecule. By optimizing the reaction conditions for the precursor synthesis, specifically the formation of the key carbazolone intermediate, the method minimizes the generation of side products at the source. 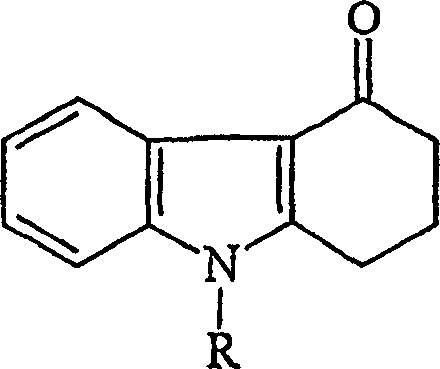 As illustrated in the structural framework of the starting materials, the precise control over the methylation and aminomethylation steps ensures a cleaner reaction profile before the final coupling occurs. The innovation extends to the final isolation step, where the use of water as a crystallization medium, combined with activated carbon treatment, provides a highly effective mechanism for scavenging trace impurities. This approach not only enhances the visual quality of the crystals but also ensures that the high-purity pharmaceutical intermediates produced meet the stringent requirements of global regulatory bodies without the need for excessive reprocessing or wasteful solvent exchanges.
As illustrated in the structural framework of the starting materials, the precise control over the methylation and aminomethylation steps ensures a cleaner reaction profile before the final coupling occurs. The innovation extends to the final isolation step, where the use of water as a crystallization medium, combined with activated carbon treatment, provides a highly effective mechanism for scavenging trace impurities. This approach not only enhances the visual quality of the crystals but also ensures that the high-purity pharmaceutical intermediates produced meet the stringent requirements of global regulatory bodies without the need for excessive reprocessing or wasteful solvent exchanges.
Mechanistic Insights into Dimethylaminomethylcarbazolone Formation and Coupling
The core of this synthetic strategy lies in the efficient construction of the carbazole backbone and its subsequent functionalization. The initial phase involves a Mannich-type reaction where methylcarbazolone is condensed with dimethylamine hydrochloride and paraformaldehyde in an acetic acid medium. This reaction environment is carefully controlled, typically maintaining temperatures between 70°C and 100°C, to facilitate the formation of the dimethylaminomethyl group at the 3-position of the carbazole ring. 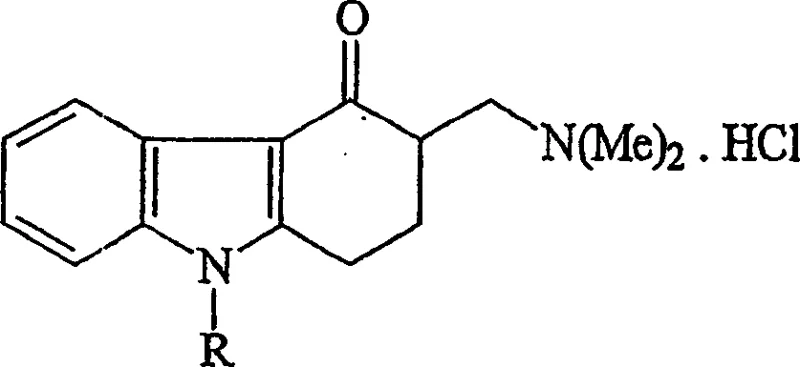 The resulting intermediate, dimethylaminomethylcarbazolone, serves as the electrophilic partner for the subsequent nucleophilic displacement by 2-methylimidazole. The mechanistic elegance of this pathway is evident in its ability to proceed with high conversion rates while suppressing the formation of the exo-methylene double bond, which typically arises from dehydration side reactions under harsher conditions. By utilizing dimethylamine hydrochloride as a stable amine source and controlling the acidity of the medium, the process stabilizes the reactive intermediates, preventing degradation pathways that lead to colored impurities. This level of mechanistic control is essential for R&D teams aiming to replicate the process at scale, as it ensures batch-to-batch consistency and minimizes the variability that often plagues multi-step organic syntheses involving heterocyclic systems.
The resulting intermediate, dimethylaminomethylcarbazolone, serves as the electrophilic partner for the subsequent nucleophilic displacement by 2-methylimidazole. The mechanistic elegance of this pathway is evident in its ability to proceed with high conversion rates while suppressing the formation of the exo-methylene double bond, which typically arises from dehydration side reactions under harsher conditions. By utilizing dimethylamine hydrochloride as a stable amine source and controlling the acidity of the medium, the process stabilizes the reactive intermediates, preventing degradation pathways that lead to colored impurities. This level of mechanistic control is essential for R&D teams aiming to replicate the process at scale, as it ensures batch-to-batch consistency and minimizes the variability that often plagues multi-step organic syntheses involving heterocyclic systems.
Furthermore, the purification mechanism employed in the final stages leverages the differential solubility properties of the target molecule versus its impurities. The conversion of the ondansetron base to its hydrochloride dihydrate salt is performed in an aqueous environment, a departure from traditional organic solvent-based crystallizations. The addition of activated carbon during this phase acts as a powerful adsorbent, selectively binding planar aromatic impurities and colored bodies that might co-precipitate with the product. The pH is meticulously adjusted to approximately 3 using hydrochloric acid, a condition that maximizes the yield of the dihydrate salt while keeping potential basic impurities in the solution phase. This selective crystallization is a critical unit operation that defines the success of the entire campaign, allowing manufacturers to achieve purity levels of 99.9% with a single or double recrystallization cycle. For technical teams, understanding this interplay between pH, solvent polarity, and adsorption is key to troubleshooting and optimizing the process for maximum throughput and minimal waste generation in a GMP environment.
How to Synthesize Ondansetron Hydrochloride Efficiently
The implementation of this improved synthesis route requires strict adherence to the specified reaction parameters to ensure the successful suppression of impurities and the attainment of the desired crystal form. The process is divided into three distinct operational phases: the preparation of the aminomethylated intermediate, the coupling with the imidazole moiety to form the base, and the final salt formation and crystallization. Each step builds upon the purity of the previous one, emphasizing the importance of in-process controls and rigorous filtration techniques. The following guide outlines the standardized approach derived from the patent specifications, designed to assist technical teams in establishing a robust manufacturing protocol that aligns with current Good Manufacturing Practices (cGMP).
- Preparation of Dimethylaminomethylcarbazolone via reflux of methylcarbazolone with dimethylamine hydrochloride and paraformaldehyde in acetic acid.
- Synthesis of Ondansetron Base by reacting the carbazolone intermediate with 2-methylimidazole followed by precipitation and washing.
- Final purification through aqueous crystallization of the hydrochloride salt using activated carbon to achieve >99.0% purity.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this improved manufacturing process offers substantial strategic benefits for procurement managers and supply chain leaders tasked with securing reliable sources of high-value APIs. The shift towards a more aqueous-based final purification step significantly reduces the dependency on volatile organic compounds (VOCs), thereby lowering the costs associated with solvent procurement, recovery, and hazardous waste disposal. This alignment with green chemistry principles not only reduces the environmental footprint of the manufacturing site but also mitigates regulatory risks associated with solvent residue limits in the final drug product. For organizations seeking a reliable ondansetron intermediate supplier, the ability to produce material with consistently low impurity profiles translates directly into reduced quality control overhead and faster release times for finished dosage forms. The simplified workflow eliminates several unit operations found in legacy processes, such as complex extractions or multiple solvent swaps, which streamlines the production timeline and enhances overall equipment effectiveness (OEE).
- Cost Reduction in Manufacturing: The economic advantages of this process are driven primarily by the optimization of raw material utilization and the simplification of downstream processing. By employing readily available reagents like paraformaldehyde and dimethylamine hydrochloride in a straightforward reflux setup, the method avoids the need for expensive or exotic catalysts that often characterize newer synthetic routes. The elimination of vacuum distillation steps in the intermediate preparation further reduces energy consumption and equipment wear, contributing to a lower cost of goods sold (COGS). Additionally, the high yield and purity achieved in the final crystallization step minimize the loss of valuable API mass during purification, ensuring that a greater proportion of the input materials are converted into saleable product. These cumulative efficiencies allow for a more competitive pricing structure without compromising on the quality attributes required for pharmaceutical applications.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the use of commodity chemicals that are widely available from multiple global vendors, reducing the risk of raw material shortages that can disrupt production schedules. The robustness of the reaction conditions, which tolerate slight variations in temperature and stoichiometry without significant degradation in product quality, ensures consistent output even in large-scale commercial reactors. This reliability is crucial for maintaining continuous supply to downstream formulation partners, particularly in the context of generic drug markets where margin pressures are high and continuity of supply is a key differentiator. Furthermore, the reduced complexity of the process facilitates easier technology transfer between manufacturing sites, enabling companies to diversify their production footprint and mitigate geopolitical or logistical risks associated with single-source manufacturing dependencies.
- Scalability and Environmental Compliance: The process is inherently scalable, having been designed with industrial application in mind, allowing for seamless transition from pilot plant batches to multi-ton commercial production campaigns. The reliance on water for the final crystallization significantly reduces the volume of organic waste streams, simplifying effluent treatment and ensuring compliance with increasingly stringent environmental regulations. The use of activated carbon, a regenerable or disposable adsorbent with a well-understood safety profile, further enhances the environmental compatibility of the process. For supply chain heads, this means fewer permitting hurdles and a smoother path to regulatory approval for new manufacturing facilities. The ability to produce high-purity material with a smaller environmental footprint also aligns with the sustainability goals of major pharmaceutical buyers, adding value beyond mere cost and availability metrics.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this advanced ondansetron synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the operational advantages for potential manufacturing partners and technical evaluators. Understanding these details is essential for making informed decisions about process adoption and supply chain integration.
Q: How does this process control the exo-methylene by-product?
A: The process utilizes a specific recrystallization protocol involving activated carbon and controlled pH adjustment during the acidification step, which effectively excludes the 1,2,3,9-tetrahydro-9-methyl-3-methylene-4H-carbazol-4-one impurity from the final crystal lattice.
Q: What are the solvent advantages in the final crystallization step?
A: Unlike conventional methods that may rely on complex organic solvent mixtures, this improved method employs water as the primary crystallization medium for the hydrochloride salt, significantly reducing solvent recovery costs and environmental impact while simplifying the isolation procedure.
Q: What purity levels can be achieved with this methodology?
A: The optimized protocol consistently yields ondansetron hydrochloride dihydrate with a purity exceeding 99.0%, with preferred embodiments achieving greater than 99.5% and up to 99.9% purity, meeting rigorous clinical standards.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ondansetron Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of purity and consistency in the production of life-saving medications like ondansetron. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in patents like CN1496350A can be successfully translated into reliable industrial reality. We operate stringent purity specifications and maintain rigorous QC labs equipped to detect trace impurities at the ppm level, guaranteeing that every batch of ondansetron hydrochloride dihydrate meets or exceeds global pharmacopeial standards. Our commitment to quality is matched by our dedication to process optimization, continuously seeking ways to enhance yield and reduce environmental impact while maintaining the highest levels of product integrity for our clients.
We invite pharmaceutical companies and contract manufacturers to engage with our technical procurement team to discuss how our capabilities align with your specific supply chain needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to our optimized manufacturing route. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project requirements, ensuring a partnership built on transparency, technical excellence, and mutual growth in the competitive global pharmaceutical market.