Advanced Crystallization Technology for High-Purity Himbacine Intermediates and Commercial Scalability
The pharmaceutical industry's relentless pursuit of potent anticoagulants has placed significant emphasis on the efficient synthesis of himbacine analogs, specifically those acting as thrombin receptor antagonists. Patent CN101910148B discloses a groundbreaking high-purity synthetic process for preparing dodecahydro-naphtho-furanyl-carbamate intermediates, which serve as the critical backbone for these life-saving medications. This technological advancement addresses a longstanding bottleneck in the manufacturing of Formula Ia compounds, where traditional methods often struggled with inconsistent purity profiles and complex purification requirements. By introducing a novel crystallization strategy that leverages specific solvent interactions, this patent provides a robust pathway for producing intermediates with impurity levels consistently below 3.0 mole percent. For R&D directors and process chemists, this represents a pivotal shift from empirical trial-and-error purification to a scientifically controlled crystallization engineering approach. The ability to reliably produce high-purity intermediates directly from the reaction mixture, without the need for multiple recrystallization cycles, fundamentally alters the economic and operational landscape of API manufacturing. This report delves deep into the mechanistic advantages and commercial implications of adopting this refined synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in US Publication 2006/0247450, relied heavily on precipitation techniques using ethanol and water mixtures to isolate the target carbamate intermediates. However, these conventional approaches exhibited severe vulnerabilities regarding product quality and process robustness. A primary deficiency was the extreme sensitivity of the final product purity to the quality of the precursor materials; even minor variations in the starting nitro compound could propagate through the synthesis, resulting in unacceptable impurity levels in the final intermediate. Furthermore, the ethanol/water system frequently led to the formation of oils rather than distinct crystals, a phenomenon known as 'oiling out,' which traps impurities within the amorphous solid and makes subsequent purification exceedingly difficult. To mitigate these issues, manufacturers were forced to implement multiple, labor-intensive recrystallization steps, which drastically reduced overall process yield and increased solvent consumption. This lack of reproducibility posed significant risks for commercial scale-up, as batch-to-batch consistency was hard to guarantee without exhaustive quality control interventions.
The Novel Approach
In stark contrast, the method disclosed in CN101910148B introduces a sophisticated solvent engineering strategy that transforms the isolation step into a highly effective purification event. Instead of relying on non-specific precipitation, this novel approach utilizes acetone or 2-methyl-tetrahydrofuran (2-Me-THF) to induce the formation of specific crystalline solvates, namely the monoacetone solvate or the mono-2-Me-THF solvate of the Formula I compound. 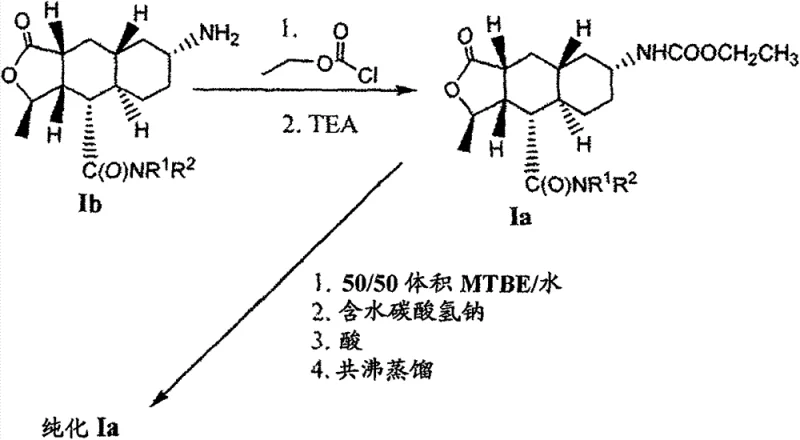 This deliberate formation of a solvated crystal lattice creates a thermodynamic barrier that effectively excludes structurally related impurities during the growth phase. The process involves a strategic solvent swap via azeotropic distillation, ensuring that the reaction medium is replaced entirely by the purification solvent before crystallization begins. This eliminates the carryover of reaction byproducts that typically plague conventional workups. Consequently, the resulting solid precipitates with exceptional purity directly from the mother liquor, often negating the need for further recrystallization and thereby streamlining the entire production workflow while enhancing the physical handling properties of the intermediate.
This deliberate formation of a solvated crystal lattice creates a thermodynamic barrier that effectively excludes structurally related impurities during the growth phase. The process involves a strategic solvent swap via azeotropic distillation, ensuring that the reaction medium is replaced entirely by the purification solvent before crystallization begins. This eliminates the carryover of reaction byproducts that typically plague conventional workups. Consequently, the resulting solid precipitates with exceptional purity directly from the mother liquor, often negating the need for further recrystallization and thereby streamlining the entire production workflow while enhancing the physical handling properties of the intermediate.
Mechanistic Insights into Solvate-Mediated Crystallization
The core innovation of this technology lies in the precise manipulation of solubility and nucleation kinetics through temperature control and solvent selection. The mechanism begins with the hydrogenation of the nitro precursor (Formula Ic) to the amine (Formula Ib), followed by carbamoylation to form the crude product. 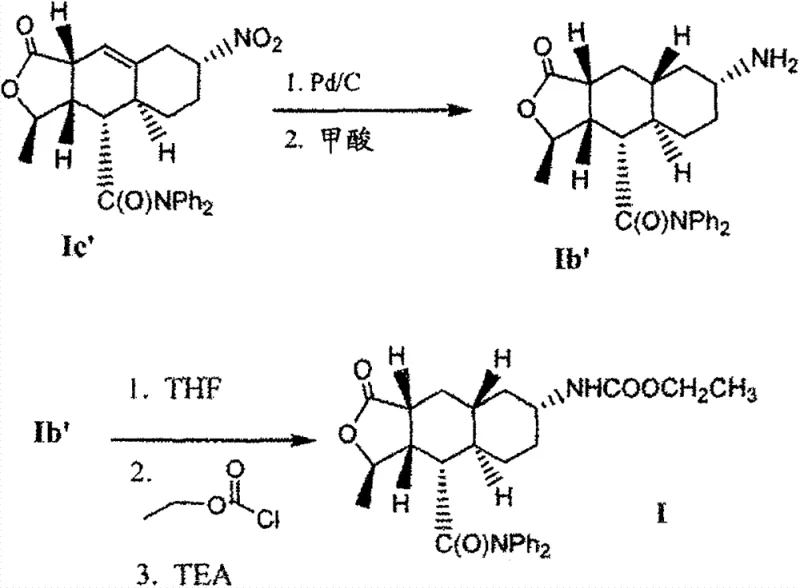 Crucially, the subsequent workup involves extracting the product into an organic phase and then performing a rigorous solvent exchange. By azeotropically distilling the mixture with acetone or 2-Me-THF, the system removes residual water and reaction solvents like THF or MTBE, which could otherwise interfere with crystal habit. The solution is then heated to a first temperature range of approximately 45°C to 60°C. At this elevated temperature, the solution becomes saturated, and seed crystals of the desired solvate begin to form. This 'seeding' phase is critical as it establishes the correct crystal polymorph. Subsequently, the mixture is cooled slowly over a period of about 3 hours to a second temperature between -5°C and +10°C. This controlled cooling rate allows for the orderly growth of the crystal lattice, maximizing the rejection of impurities into the mother liquor. The specific interaction between the carbamate molecule and the solvent molecules (acetone or 2-Me-THF) stabilizes the crystal structure, preventing the disorder that leads to oil formation. This thermodynamic stability ensures that the isolated solid maintains a consistent stoichiometry and purity profile, regardless of minor fluctuations in the upstream reaction conditions.
Crucially, the subsequent workup involves extracting the product into an organic phase and then performing a rigorous solvent exchange. By azeotropically distilling the mixture with acetone or 2-Me-THF, the system removes residual water and reaction solvents like THF or MTBE, which could otherwise interfere with crystal habit. The solution is then heated to a first temperature range of approximately 45°C to 60°C. At this elevated temperature, the solution becomes saturated, and seed crystals of the desired solvate begin to form. This 'seeding' phase is critical as it establishes the correct crystal polymorph. Subsequently, the mixture is cooled slowly over a period of about 3 hours to a second temperature between -5°C and +10°C. This controlled cooling rate allows for the orderly growth of the crystal lattice, maximizing the rejection of impurities into the mother liquor. The specific interaction between the carbamate molecule and the solvent molecules (acetone or 2-Me-THF) stabilizes the crystal structure, preventing the disorder that leads to oil formation. This thermodynamic stability ensures that the isolated solid maintains a consistent stoichiometry and purity profile, regardless of minor fluctuations in the upstream reaction conditions.
Furthermore, the impurity control mechanism is deeply rooted in the differential solubility of the target compound versus its byproducts in the chosen solvating media. Impurities that might co-precipitate in an ethanol/water system remain highly soluble in the acetone or 2-Me-THF mother liquor at the lower crystallization temperatures. The patent data indicates that this method can reduce impurity levels by a factor of ten compared to prior art, achieving levels below 2.0 mole percent in optimized runs. This high level of purification is achieved not through additional processing steps, but through the intrinsic selectivity of the crystallization process itself. For process chemists, this means that the burden of purification is shifted from downstream separation units to the crystallization reactor, simplifying the plant design and reducing the footprint required for production. The ability to consistently produce a crystalline solvate also improves the filtration and drying characteristics of the material, reducing cycle times and energy consumption during the isolation phase.
How to Synthesize Dodecahydro-naphtho-furanyl-carbamate Efficiently
The synthesis of this critical pharmaceutical intermediate requires strict adherence to the patented solvent exchange and temperature profiling protocols to ensure the formation of the high-purity solvate. The process integrates the chemical transformation with the purification step, creating a seamless operation that minimizes material handling. Operators must carefully monitor the azeotropic distillation to ensure complete removal of the initial reaction solvents before initiating the cooling ramp.
- Hydrogenation of the nitro precursor (Formula Ic) using palladium on carbon in a THF/water mixture, followed by pH adjustment and extraction to isolate the amine intermediate.
- Reaction of the amine intermediate with ethyl chloroformate in the presence of a base like triethylamine to form the crude carbamate product in an organic phase.
- Solvent exchange via azeotropic distillation to replace the reaction solvent with acetone or 2-methyl-tetrahydrofuran, followed by controlled cooling crystallization to precipitate the pure solvate form.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this advanced crystallization technology offers substantial strategic benefits that extend far beyond simple yield improvements. The primary advantage lies in the drastic simplification of the manufacturing process, which directly translates to cost reduction in API manufacturing. By eliminating the need for multiple recrystallization cycles, the process significantly reduces solvent consumption, waste generation, and labor hours associated with reprocessing off-spec batches. This efficiency gain allows for a more competitive pricing structure without compromising on the stringent quality standards required for cardiovascular therapeutics. Moreover, the robustness of the process against feedstock variability means that suppliers can source raw materials from a broader range of vendors without risking production stoppages due to purity issues. This flexibility enhances supply chain resilience, ensuring continuous availability of this critical intermediate even during periods of raw material scarcity.
- Cost Reduction in Manufacturing: The elimination of redundant purification steps creates a leaner manufacturing workflow that significantly lowers the cost of goods sold. Since the crystallization step itself acts as a high-efficiency purification unit, the capital expenditure required for additional filtration and drying equipment is minimized. Furthermore, the reduced solvent usage aligns with green chemistry principles, lowering the costs associated with solvent recovery and hazardous waste disposal. This operational efficiency allows manufacturers to allocate resources towards capacity expansion rather than waste management, driving long-term profitability.
- Enhanced Supply Chain Reliability: The insensitivity of this process to minor variations in precursor purity mitigates the risk of batch failures, which are a common cause of supply disruptions in the pharmaceutical sector. A more predictable production schedule enables better inventory planning and shorter lead times for high-purity pharmaceutical intermediates. Suppliers utilizing this technology can offer more reliable delivery commitments, fostering stronger partnerships with downstream API manufacturers who depend on just-in-time delivery models to maintain their own production schedules.
- Scalability and Environmental Compliance: The use of solvents like 2-methyl-tetrahydrofuran, which is derived from renewable resources and has a favorable environmental profile, supports corporate sustainability goals. The process is inherently scalable, as the crystallization parameters (temperature and cooling rate) can be precisely controlled in large-scale reactors using standard automation systems. This ease of scale-up facilitates the commercial scale-up of complex pharmaceutical intermediates from pilot plant quantities to multi-ton annual production volumes without the need for extensive process re-optimization, ensuring a smooth transition from development to commercial supply.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this high-purity synthesis method. These insights are derived directly from the experimental data and process descriptions found in the patent literature, providing a clear understanding of the technology's capabilities.
Q: Why is the new crystallization method superior to the prior ethanol/water precipitation technique?
A: The prior art method often resulted in the formation of oils or required multiple recrystallization steps to achieve acceptable purity, making it highly sensitive to precursor quality. The novel method utilizes specific solvate formation (acetone or 2-Me-THF) which inherently excludes impurities during crystal lattice formation, consistently delivering purity levels below 3.0 mole percent without extensive reprocessing.
Q: What are the critical process parameters for ensuring high yield in this synthesis?
A: Critical parameters include the precise control of temperature during the solvent exchange phase, maintaining the first crystallization temperature between 45°C and 60°C to initiate nucleation, and subsequently cooling to a second temperature between -5°C and +10°C over a controlled period of approximately 3 hours to maximize crystal growth and impurity rejection.
Q: How does this process impact the scalability of thrombin receptor antagonist production?
A: By eliminating the need for multiple recrystallization cycles and reducing the sensitivity to feedstock purity, the process significantly simplifies the manufacturing workflow. This robustness allows for easier commercial scale-up, reduces batch-to-batch variability, and minimizes the risk of production delays associated with purification failures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dodecahydro-naphtho-furanyl-carbamate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of next-generation anticoagulants depends on the availability of high-quality intermediates produced via robust and scalable processes. Our technical team has extensively analyzed the crystallization technologies described in CN101910148B and possesses the expertise to implement these advanced purification strategies effectively. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory synthesis to industrial manufacturing is seamless and efficient. Our facilities are equipped with state-of-the-art reactors capable of precise temperature control and azeotropic distillation, which are essential for executing the solvent exchange and crystallization steps required by this patent. We adhere to stringent purity specifications and operate rigorous QC labs to guarantee that every batch of dodecahydro-naphtho-furanyl-carbamate meets the exacting standards necessary for thrombin receptor antagonist synthesis.
We invite potential partners to engage with our technical procurement team to discuss how this innovative synthesis route can optimize your supply chain. By leveraging our capabilities, you can secure a Customized Cost-Saving Analysis that quantifies the potential reductions in manufacturing costs and lead times specific to your project requirements. We encourage you to contact us to request specific COA data and route feasibility assessments, allowing you to make informed decisions about integrating this high-purity intermediate into your API production strategy. Together, we can accelerate the delivery of vital cardiovascular therapies to patients worldwide.