Advanced Synthesis of Chiral Cyclic Beta-Amino Aryl Butyric Acid Derivatives for Pharmaceutical Applications
Advanced Synthesis of Chiral Cyclic Beta-Amino Aryl Butyric Acid Derivatives for Pharmaceutical Applications
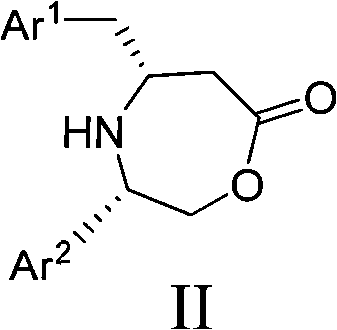
The pharmaceutical industry continuously demands high-purity chiral intermediates to ensure the efficacy and safety of final drug products. Patent CN102212041A introduces a groundbreaking methodology for the preparation of chiral cyclic β-aminoaryl butyric acid derivatives, specifically designated as structural formula II. These compounds serve as critical building blocks in the synthesis of various chiral drugs, offering a robust alternative to traditional synthetic routes. The innovation lies in its ability to achieve exceptional optical purity exceeding 99.0% ee through a streamlined process that combines condensation with asymmetric induced reduction. This technical advancement addresses long-standing challenges in scalability and cost-efficiency, making it a highly attractive option for large-scale industrial production. By leveraging chiral amino alcohols as auxiliary groups, the method ensures precise stereocontrol without relying on prohibitively expensive transition metal catalysts.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral β-aminoaryl butyric acid derivatives has relied heavily on homogeneous asymmetric hydrogenation or chiral resolution techniques, both of which present significant drawbacks for commercial manufacturing. Homogeneous catalysis often necessitates the use of precious metal complexes, such as Ru-(S)-BINAP or rhodium ligands, which are not only costly but also difficult to recover and recycle, leading to substantial waste generation. Furthermore, these reactions typically require stringent anaerobic conditions and specialized equipment to prevent catalyst deactivation, complicating the operational workflow and increasing capital expenditure. Chiral resolution methods, while effective in separating enantiomers, suffer from a theoretical maximum yield of 50%, meaning half of the synthesized material is discarded or requires complex racemization processes, thereby negatively impacting both economic efficiency and environmental sustainability.
The Novel Approach
In stark contrast, the novel approach detailed in the patent utilizes a chiral auxiliary induction strategy that circumvents the need for expensive metal catalysts and harsh reaction environments. By condensing aryl acetoacetates with readily available chiral amino alcohols, such as (S)-phenylglycinol, the process creates a chiral environment that directs the subsequent reduction with high fidelity. This method operates under much milder conditions, often at room temperature or with simple heating, and does not demand the rigorous exclusion of oxygen required by sensitive metal catalysts. The result is a synthesis pathway that is not only simpler to operate but also inherently more scalable, allowing manufacturers to produce high-purity intermediates with significantly reduced overhead costs and improved process safety profiles.
Mechanistic Insights into Asymmetric Induced Reduction
The core of this synthetic strategy involves a multi-step transformation that begins with the condensation of an aryl or heteroaryl acetoacetate (III) with a chiral amino alcohol to form an intermediate (IV). This initial step establishes the chiral framework necessary for stereochemical control. Following condensation, the intermediate undergoes cyclization, often facilitated by a base like sodium hydride, to form a cyclic enamine or imine structure (V). This cyclic intermediate locks the conformation of the molecule, ensuring that the subsequent reduction occurs from a specific face of the double bond. The rigidity of the seven-membered ring system in intermediate V is crucial for transmitting the chiral information from the auxiliary group to the newly formed stereocenter.
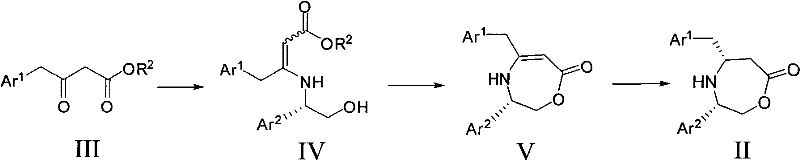
The final and most critical step is the asymmetric induced reduction of the cyclic intermediate V to yield the target chiral derivative II. This reduction is typically performed using hydride sources such as sodium cyanoborohydride or sodium borohydride in the presence of an acid like acetic acid. The chiral auxiliary group sterically hinders one face of the unsaturated bond, forcing the hydride attack to occur exclusively from the less hindered side. This steric guidance results in the formation of a single enantiomer with high optical purity, often eliminating the need for further purification steps like recrystallization. The ability to tune the stereochemistry by simply switching between (S)- or (R)-configured amino alcohols provides manufacturers with flexible access to either enantiomer of the final product, catering to diverse drug development needs.
How to Synthesize Chiral Cyclic Beta-Amino Aryl Butyric Acid Derivatives Efficiently
The synthesis protocol outlined in the patent offers a clear and reproducible pathway for generating these valuable intermediates. The process begins with the preparation of the starting acetoacetate, followed by condensation with the chiral amine, cyclization, and finally reduction. Each step has been optimized to maximize yield and purity, with specific attention paid to solvent selection and temperature control. For instance, the use of tetrahydrofuran (THF) as a solvent during the condensation and cyclization steps ensures good solubility of reactants, while the reduction step benefits from a mixture of dichloromethane and dioxane to facilitate smooth hydride transfer. Detailed standard operating procedures for scaling this route from laboratory to pilot plant are essential for maintaining consistency.
- Condense aryl or heteroaryl acetoacetate (III) with a chiral amino alcohol, such as (S)-phenylglycinol, to form intermediate IV.
- Cyclize intermediate IV under basic conditions using a hydride source like NaH to obtain the cyclic intermediate V.
- Perform asymmetric induced reduction on cyclic intermediate V using a reducing agent like sodium cyanoborohydride to yield the final chiral derivative II.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis route offers tangible benefits that extend beyond mere technical feasibility. The elimination of precious metal catalysts represents a direct reduction in raw material costs, as there is no longer a need to purchase expensive ruthenium or rhodium complexes or invest in their recovery systems. Additionally, the simplified operational requirements mean that production can be carried out in standard chemical reactors without the need for specialized high-pressure hydrogenation equipment or inert atmosphere gloveboxes, drastically lowering capital investment barriers. The high optical purity achieved directly from the reaction minimizes downstream processing steps, reducing solvent consumption, energy usage, and labor hours associated with recrystallization and purification.
- Cost Reduction in Manufacturing: The replacement of homogeneous metal catalysis with organic chiral auxiliaries removes the dependency on volatile precious metal markets and expensive ligand synthesis. This shift leads to substantial cost savings in raw material procurement and waste disposal, as heavy metal contamination is no longer a concern requiring costly remediation. Furthermore, the high yields reported in the examples, such as the 94% yield in the preparation of intermediate IIIa and 78% in the final reduction step, contribute to a more efficient use of starting materials, lowering the overall cost per kilogram of the final active pharmaceutical ingredient.
- Enhanced Supply Chain Reliability: The reagents used in this process, including aryl acetoacetates and chiral amino alcohols like phenylglycinol, are commercially available in bulk quantities from multiple global suppliers. This diversity in sourcing mitigates the risk of supply disruptions that often plague specialized catalyst-dependent processes. The robustness of the reaction conditions also means that production schedules are less likely to be delayed by equipment failures or environmental control issues, ensuring a steady and reliable flow of intermediates to meet tight drug development timelines.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing common organic solvents and standard unit operations that are easily transferred from benchtop to multi-ton production. From an environmental perspective, the absence of heavy metals simplifies effluent treatment and reduces the regulatory burden associated with hazardous waste disposal. The high atom economy and reduced solvent usage due to fewer purification steps align with green chemistry principles, helping companies meet increasingly stringent environmental regulations and sustainability goals without compromising on product quality.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. Understanding these details is crucial for R&D teams evaluating the feasibility of integrating this route into their existing manufacturing pipelines. The answers are derived directly from the experimental data and claims presented in the patent documentation, ensuring accuracy and reliability for decision-making purposes.
Q: What is the primary advantage of this synthesis method over homogeneous asymmetric hydrogenation?
A: This method avoids the use of expensive precious metal catalysts like Ruthenium or Rhodium and eliminates the need for strict anaerobic conditions, significantly reducing operational complexity and cost.
Q: What optical purity can be achieved with this process?
A: The patented process consistently achieves an optical purity greater than 99.0% ee, minimizing the need for extensive recrystallization steps often required in other methods.
Q: Which reducing agents are compatible with this synthesis route?
A: The process supports a wide range of reducing agents including sodium cyanoborohydride, sodium borohydride, and lithium aluminum hydride, with sodium cyanoborohydride being the preferred choice for optimal results.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Cyclic Beta-Amino Aryl Butyric Acid Derivatives Supplier
NINGBO INNO PHARMCHEM stands at the forefront of custom synthesis and contract manufacturing, possessing the technical expertise to translate complex patent methodologies like CN102212041A into commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instruments to verify the optical purity and chemical integrity of every batch, guaranteeing that our chiral intermediates meet the highest industry standards.
We invite you to collaborate with us to optimize your supply chain and reduce your manufacturing costs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific project requirements. We are ready to provide specific COA data and comprehensive route feasibility assessments to demonstrate how our capabilities can accelerate your drug development timeline and enhance your competitive edge in the global market.