Advanced Manufacturing of Beta-3 Agonist Intermediates via Streamlined Epoxide Ring-Opening
Advanced Manufacturing of Beta-3 Agonist Intermediates via Streamlined Epoxide Ring-Opening
The pharmaceutical landscape for metabolic disorders continues to evolve, driven by the urgent need for effective treatments for obesity and type 2 diabetes. Central to this therapeutic area are arylethanoldiamine derivatives, which function as potent agonists at atypical beta-adrenoceptors, specifically the beta-3 subtype. The patent CN1487916A discloses a groundbreaking improvement in the preparation of these critical compounds, addressing long-standing challenges in synthetic efficiency and scalability. This technical insight report analyzes the proprietary methodology outlined in the patent, highlighting its potential to redefine the supply chain for high-purity pharmaceutical intermediates. By leveraging a novel epoxide ring-opening strategy, the disclosed process achieves superior regioselectivity and yield compared to conventional routes. 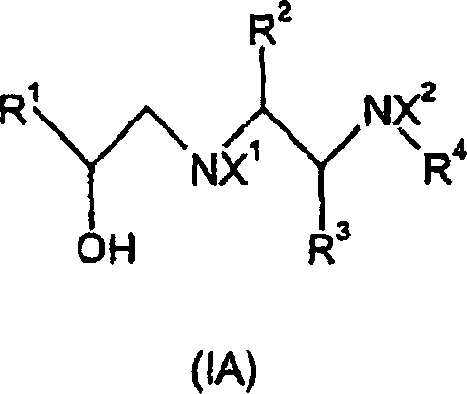 For R&D directors and procurement specialists, understanding the nuances of this chemistry is essential for securing a reliable arylethanoldiamine supplier capable of meeting stringent quality standards. The innovation lies not just in the final molecule, but in the robust construction of the carbon-nitrogen backbone, which minimizes impurity profiles and simplifies downstream purification.
For R&D directors and procurement specialists, understanding the nuances of this chemistry is essential for securing a reliable arylethanoldiamine supplier capable of meeting stringent quality standards. The innovation lies not just in the final molecule, but in the robust construction of the carbon-nitrogen backbone, which minimizes impurity profiles and simplifies downstream purification.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex arylethanoldiamine scaffolds has been plagued by inefficiencies that drive up costs and extend lead times for high-purity pharmaceutical intermediates. Traditional pathways often rely on multi-step sequences involving protecting group manipulations that add unnecessary complexity to the manufacturing process. A significant bottleneck in prior art methods is the lack of regiocontrol during the formation of the ethanolamine side chain, frequently resulting in mixtures of regioisomers that are difficult and expensive to separate. Furthermore, many established routes necessitate the use of hazardous or expensive reagents, such as organoboron compounds, in the final coupling stages, which introduces environmental liabilities and complicates waste management. The cumulative effect of these drawbacks is a process with suboptimal atom economy and lower overall yields, making commercial scale-up of complex pharmaceutical intermediates economically challenging. Additionally, the reliance on cryogenic conditions or exotic catalysts in older methodologies often limits the throughput capacity of production facilities, creating supply chain vulnerabilities for drug developers.
The Novel Approach
In stark contrast, the method disclosed in CN1487916A offers a paradigm shift towards leaner and more sustainable manufacturing. The core innovation involves the direct reaction of a specific imidazoline intermediate with an epoxide derivative under thermal conditions, effectively bypassing the need for cumbersome protection-deprotection cycles. This approach drastically simplifies the synthetic route, reducing the total number of unit operations required to reach the final active pharmaceutical ingredient precursor. By eliminating the need for boron-containing chemicals in the key bond-forming step, the process not only reduces raw material costs but also aligns with green chemistry principles by minimizing toxic waste generation. The patent emphasizes that this new route provides higher yields than prior art methods, directly translating to better resource utilization and lower cost of goods sold. Moreover, the high regioselectivity of the epoxide ring-opening ensures a cleaner reaction profile, significantly reducing the burden on purification teams and enhancing the overall purity of the final product. This streamlined methodology represents a substantial advancement in cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Thermal Epoxide Ring-Opening
The heart of this improved process lies in the mechanistic elegance of the thermal coupling between the imidazoline species and the chiral epoxide. As illustrated in the reaction scheme, the nucleophilic nitrogen of the imidazoline ring attacks the less hindered carbon of the epoxide, driven by the elevated thermal energy provided by the reaction medium.  The patent specifies that this transformation is optimally conducted at temperatures ranging from 100°C to 150°C, with a preferred window of 100°C to 120°C. This thermal activation is crucial for overcoming the activation energy barrier of the ring-opening without the need for aggressive Lewis acids that might compromise the stereochemical integrity of the chiral center. The use of high-boiling aromatic solvents such as toluene or xylene is particularly advantageous, as they provide a homogeneous phase that facilitates efficient heat transfer and mass transport between the reactants. From an impurity control perspective, the inherent selectivity of this thermal pathway minimizes the formation of bis-alkylated byproducts or polymeric tars that are common in acid-catalyzed variants. The stability of the imidazoline ring under these conditions is also a key factor, allowing the reaction to proceed to completion while maintaining the structural fidelity of the heterocyclic core. This mechanistic robustness is what enables the process to be scaled reliably from kilogram to multi-ton quantities.
The patent specifies that this transformation is optimally conducted at temperatures ranging from 100°C to 150°C, with a preferred window of 100°C to 120°C. This thermal activation is crucial for overcoming the activation energy barrier of the ring-opening without the need for aggressive Lewis acids that might compromise the stereochemical integrity of the chiral center. The use of high-boiling aromatic solvents such as toluene or xylene is particularly advantageous, as they provide a homogeneous phase that facilitates efficient heat transfer and mass transport between the reactants. From an impurity control perspective, the inherent selectivity of this thermal pathway minimizes the formation of bis-alkylated byproducts or polymeric tars that are common in acid-catalyzed variants. The stability of the imidazoline ring under these conditions is also a key factor, allowing the reaction to proceed to completion while maintaining the structural fidelity of the heterocyclic core. This mechanistic robustness is what enables the process to be scaled reliably from kilogram to multi-ton quantities.
Following the coupling step, the process incorporates a highly efficient hydrolysis stage to unveil the final functional groups. The intermediate compound, typically an ester or protected amine, is subjected to hydrolysis using Group 1 or Group 2 metal hydroxides, such as sodium hydroxide or potassium hydroxide, in an aqueous alkanol mixture. This saponification step is designed to be exhaustive yet selective, ensuring that the sensitive ethanolamine linkage remains intact while cleaving the ester moieties to generate the free carboxylic acid. The patent notes that this hydrolysis can be performed under reflux conditions for at least 4 hours, providing ample time for complete conversion. The choice of methanol or ethanol as the co-solvent enhances the solubility of the organic intermediate in the aqueous base, preventing phase separation issues that could lead to incomplete reaction. Furthermore, the ability to perform partial hydrolysis allows for the isolation of specific mono-protected derivatives if required for downstream diversification, adding flexibility to the synthetic platform. This controlled deprotection strategy is vital for managing the impurity profile, as it avoids the harsh acidic conditions that could lead to racemization of the chiral alcohol center.
How to Synthesize Arylethanoldiamine Derivatives Efficiently
Implementing this synthesis requires precise control over reaction parameters to maximize the benefits of the patented route. The process begins with the preparation of the imidazoline precursor, which can be generated in situ or isolated as a stable salt, followed by the addition of the chiral epoxide. The reaction mixture is then heated to the specified thermal window, monitored closely to ensure the temperature does not exceed the upper limit which could induce degradation.
- Prepare the imidazoline intermediate (Formula IB) by reacting an epoxide derivative with an imidazoline precursor under elevated temperature (100-150°C) in aromatic solvents.
- Perform hydrolysis of the intermediate using Group 1 or 2 metal hydroxides (e.g., NaOH) in aqueous alkanol solutions to reveal the free amine and carboxylic acid functionalities.
- Isolate the final arylethanoldiamine product through controlled crystallization or salt formation, ensuring high purity suitable for pharmaceutical applications.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this improved synthesis route offers tangible strategic benefits beyond mere technical superiority. The simplification of the synthetic pathway directly correlates to a reduction in manufacturing complexity, which is a primary driver of cost volatility in the fine chemical sector. By removing expensive and hazardous reagents from the bill of materials, the process inherently lowers the raw material cost base, providing a buffer against market fluctuations in specialty chemical pricing.  The elimination of boron reagents, in particular, removes a significant cost center associated with both the purchase of these materials and the subsequent disposal of boron-containing waste streams. This leads to substantial cost savings in waste treatment and regulatory compliance, enhancing the overall economic viability of the project. Furthermore, the use of commodity solvents like toluene and methanol ensures that the process is not dependent on niche supply chains that might be prone to disruption. The robustness of the thermal coupling step means that the reaction is less sensitive to minor variations in feedstock quality, thereby improving batch-to-batch consistency and reducing the rate of rejected lots. These factors combined create a more resilient supply chain capable of sustaining long-term commercial production without the frequent interruptions associated with fragile, multi-step syntheses.
The elimination of boron reagents, in particular, removes a significant cost center associated with both the purchase of these materials and the subsequent disposal of boron-containing waste streams. This leads to substantial cost savings in waste treatment and regulatory compliance, enhancing the overall economic viability of the project. Furthermore, the use of commodity solvents like toluene and methanol ensures that the process is not dependent on niche supply chains that might be prone to disruption. The robustness of the thermal coupling step means that the reaction is less sensitive to minor variations in feedstock quality, thereby improving batch-to-batch consistency and reducing the rate of rejected lots. These factors combined create a more resilient supply chain capable of sustaining long-term commercial production without the frequent interruptions associated with fragile, multi-step syntheses.
- Cost Reduction in Manufacturing: The streamlined nature of the process significantly reduces the number of isolation and purification steps required, which are typically the most expensive phases of chemical manufacturing. By achieving higher yields in the key coupling step, the amount of starting material needed per kilogram of final product is decreased, directly improving the material efficiency of the plant. The avoidance of cryogenic conditions and exotic catalysts further reduces utility costs and capital expenditure on specialized equipment. Consequently, the overall cost of goods is optimized, allowing for more competitive pricing strategies in the global marketplace.
- Enhanced Supply Chain Reliability: The reliance on widely available, commodity-grade chemicals ensures that the supply chain is not vulnerable to the shortages often seen with specialized reagents. The robustness of the reaction conditions allows for flexible scheduling and easier scale-up, meaning that production capacity can be ramped up quickly to meet surging demand without extensive re-validation. This reliability is critical for pharmaceutical partners who require guaranteed continuity of supply to maintain their own clinical and commercial timelines. The simplified logistics of handling fewer distinct chemical inputs also reduces the administrative burden on procurement teams.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing standard reactor configurations and workup procedures that are easily transferable from pilot plant to full commercial scale. The reduction in toxic byproducts and the elimination of heavy metal or boron waste streams significantly simplify the environmental permitting process and reduce the long-term liability associated with chemical manufacturing. This alignment with green chemistry principles not only meets current regulatory standards but also future-proofs the manufacturing site against increasingly stringent environmental laws. The ability to recycle solvents like toluene further enhances the sustainability profile of the operation.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. Understanding these details is crucial for stakeholders evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What is the primary advantage of the process described in CN1487916A?
A: The primary advantage is the significant improvement in yield and regioselectivity during the epoxide ring-opening step, coupled with a reduction in the number of synthetic steps compared to prior art methods.
Q: Does this method require expensive boron-containing reagents?
A: No, one of the key environmental and cost benefits of this invention is the elimination of the need for boron-containing chemicals in the final assembly of the arylethanoldiamine scaffold.
Q: What are the typical reaction conditions for the key coupling step?
A: The key coupling reaction is typically conducted at elevated temperatures ranging from 100°C to 150°C, preferably using toluene or xylene as the solvent to facilitate the ring-opening mechanism.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Arylethanoldiamine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of robust synthetic routes in the development of next-generation metabolic therapeutics. Our team of expert chemists has extensively analyzed the methodology disclosed in CN1487916A and possesses the technical capability to execute this process with precision and efficiency. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from clinical trials to market launch. Our state-of-the-art facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications for every batch of arylethanoldiamine derivatives we produce. We understand that consistency is key in pharmaceutical manufacturing, and our quality management systems are designed to deliver products that meet the highest international standards.
We invite you to collaborate with us to leverage this advanced technology for your drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and timeline. Please contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our expertise can accelerate your path to commercial success while optimizing your supply chain economics.