Scalable Synthesis of Imidazopyridine P2X3 Antagonist Intermediates via Optimized Acylation and Cyclization
The pharmaceutical landscape for treating chronic cough and pain is rapidly evolving, with P2X3 receptor antagonists emerging as a highly promising therapeutic class. Patent CN114805340A details a sophisticated and robust preparation method for imidazopyridine compounds and their critical intermediates, specifically targeting the modulation of P2X3 receptors. These receptors, widely expressed in nervous and immune systems, play a pivotal role in nociception and cough reflex hypersensitivity. The disclosed technology provides a streamlined pathway to access complex intermediates of Formula I, II, and III, which serve as the structural backbone for potent antagonists. By leveraging advanced organometallic techniques and transition metal catalysis, this process addresses key challenges in synthesizing polyfluorinated aromatic systems, offering a reliable solution for the production of high-purity pharmaceutical intermediates.
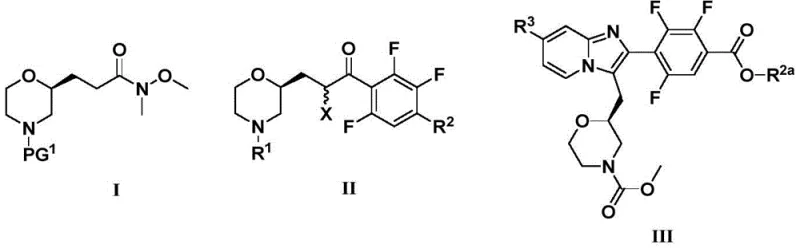
For R&D directors and process chemists, the significance of this patent lies in its ability to construct the imidazopyridine core with high fidelity. The method avoids common pitfalls associated with polyfluorinated substrates, such as non-selective substitution or decomposition under harsh conditions. Instead, it utilizes a sequence of well-controlled reactions, including lithiation, acylation, carbonylation, and cyclization. This approach not only enhances the overall yield but also simplifies the purification profile, which is crucial for meeting the stringent quality standards required for clinical-grade active pharmaceutical ingredients (APIs). The versatility of the route allows for the introduction of various substituents, enabling the rapid generation of analog libraries for structure-activity relationship (SAR) studies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic routes for constructing polyfluorinated ketone intermediates often suffer from significant drawbacks that impede efficient manufacturing. Conventional Friedel-Crafts acylations on highly fluorinated benzenes frequently result in mixtures of regioisomers due to the similar electronic environments of the fluorine-substituted positions. Separating these isomers is notoriously difficult and costly, often requiring multiple chromatographic steps that drastically reduce overall throughput. Furthermore, older methods for installing the ester functionality on the aromatic ring might involve harsh hydrolysis conditions or multi-step protection-deprotection sequences that generate excessive waste. The use of unstable organometallic reagents without proper stabilization can also lead to safety hazards and inconsistent batch-to-batch reproducibility, making these legacy processes unsuitable for modern GMP production environments.
The Novel Approach
The methodology outlined in CN114805340A represents a paradigm shift by employing a Weinreb amide strategy coupled with directed ortho-lithiation. By reacting a protected morpholine Weinreb amide with a specifically generated aryl lithium species, the process ensures mono-acylation with exceptional regioselectivity. This eliminates the formation of tertiary alcohol byproducts that typically arise from over-addition of organometallic reagents to ketones. Additionally, the integration of a palladium-catalyzed carbonylation step allows for the direct conversion of an aryl bromide to a methyl ester under mild pressure (40-50 psi CO). This telescoped approach reduces the number of unit operations and avoids the use of toxic stoichiometric oxidants. The final cyclization step utilizes a simple thermal reaction with substituted 2-aminopyridines, demonstrating a clear advantage in operational simplicity and scalability compared to traditional condensation methods.
Mechanistic Insights into LDA-Mediated Acylation and Pd-Catalyzed Carbonylation
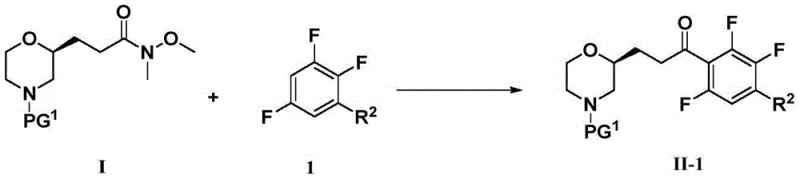
The core of this synthetic strategy relies on the precise generation of a nucleophilic aryl species from a polyfluorinated benzene derivative. In Step 1 of the intermediate preparation, lithium diisopropylamide (LDA) acts as a strong, non-nucleophilic base to deprotonate the aromatic ring at the position ortho to the fluorine atoms, which is activated by the electron-withdrawing nature of the halogens. This lithiation occurs at controlled low temperatures, typically between -10 to 0°C, to prevent side reactions such as benzyne formation or elimination. The resulting aryl lithium intermediate is then immediately trapped by the Weinreb amide (Formula I-3). The N-methoxy-N-methylamide moiety is critical here; it forms a stable tetrahedral intermediate with the organolithium reagent that does not collapse to the ketone until acidic workup. This mechanistic feature effectively prevents the second addition of the aryl lithium to the newly formed ketone, ensuring high selectivity for the desired mono-acylated product.
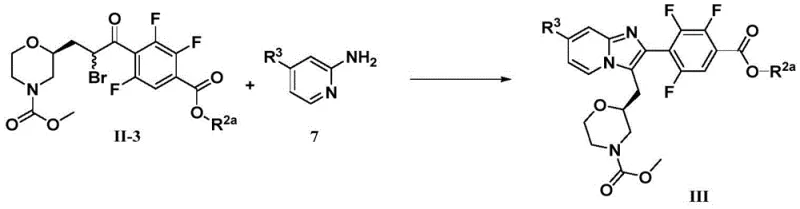
Following the acylation, the installation of the ester group is achieved through a sophisticated palladium-catalyzed carbonylation. In this step, the aryl bromide intermediate undergoes oxidative addition with a Pd(0) catalyst, specifically Pd(dppf)Cl2, which is known for its stability and activity in coupling reactions involving sterically hindered substrates. Under a carbon monoxide atmosphere of approximately 45 psi, the aryl-palladium species inserts CO to form an acyl-palladium complex. Subsequent nucleophilic attack by methanol, facilitated by an organic base like triethylamine, releases the methyl ester product and regenerates the active catalyst. This transformation is highly atom-economical and avoids the need for pre-functionalized boronic acids or tin reagents used in Suzuki or Stille couplings. The final cyclization to form the imidazopyridine ring involves an SN2-type displacement of an alpha-bromo ketone by the exocyclic amine of a 2-aminopyridine, followed by dehydration, driven by the aromaticity of the newly formed fused heterocyclic system.
How to Synthesize Imidazopyridine Intermediates Efficiently
The synthesis of these high-value intermediates requires careful attention to reaction parameters, particularly temperature control and reagent stoichiometry, to maximize yield and purity. The process begins with the preparation of the chiral morpholine Weinreb amide, followed by the critical acylation step described above. Subsequent transformations involve carbonylation, deprotection, re-protection if necessary, alpha-bromination, and finally, the heterocyclic ring closure. Each step has been optimized to use commercially available solvents like THF, DCM, and acetonitrile, facilitating easy solvent recovery and recycling. For detailed operational procedures, including specific molar ratios, addition rates, and workup protocols, please refer to the standardized synthesis guide below which encapsulates the critical process parameters defined in the patent documentation.
- Perform regioselective acylation by reacting a protected morpholine Weinreb amide with a polyfluorinated aryl lithium species generated in situ using LDA at low temperatures.
- Execute a palladium-catalyzed carbonylation reaction under carbon monoxide pressure to convert the aryl bromide intermediate into the corresponding methyl ester.
- Complete the synthesis via alpha-bromination of the ketone followed by cyclization with a substituted 2-aminopyridine to form the imidazo[1,2-a]pyridine core.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, the adoption of this patented process offers substantial strategic benefits that extend beyond mere technical feasibility. The reliance on commodity chemicals such as polyfluorobenzenes, morpholine derivatives, and standard palladium catalysts ensures a resilient supply chain that is less susceptible to the volatility associated with exotic or custom-synthesized starting materials. The robustness of the reaction conditions, particularly the tolerance for mild temperatures and pressures in the carbonylation and cyclization steps, significantly lowers the barrier for technology transfer from laboratory to pilot and commercial plant scales. This reduces the risk of production delays and ensures a consistent supply of critical intermediates for downstream API manufacturing.
- Cost Reduction in Manufacturing: The process achieves cost efficiency primarily through improved selectivity and reduced waste generation. By utilizing the Weinreb amide strategy, the formation of difficult-to-remove over-addition byproducts is virtually eliminated, which drastically cuts down on purification costs and solvent consumption. Furthermore, the direct carbonylation step replaces multi-step sequences that would otherwise require expensive reagents and generate stoichiometric amounts of salt waste. The use of catalytic amounts of palladium, which can potentially be recovered and recycled, further optimizes the cost structure, making the overall manufacturing process economically competitive for large-scale production.
- Enhanced Supply Chain Reliability: The synthetic route is designed with supply chain continuity in mind, utilizing reagents that are globally sourced and readily available in bulk quantities. The avoidance of cryogenic conditions (below -40°C) for extended periods reduces the energy burden on manufacturing facilities and minimizes the dependency on specialized cooling infrastructure. This operational flexibility allows for production in a wider range of geographic locations, mitigating regional supply risks. Additionally, the stability of the intermediates, such as the bromo-ketone and the protected morpholine derivatives, allows for safe storage and transportation, providing buffer stock options that enhance overall supply security.
- Scalability and Environmental Compliance: The methodology aligns well with green chemistry principles, contributing to a more sustainable manufacturing footprint. The high atom economy of the carbonylation reaction and the efficient use of solvents reduce the E-factor (mass of waste per mass of product). The process avoids the use of heavy metal stoichiometric oxidants or highly toxic reagents, simplifying wastewater treatment and regulatory compliance. The scalability is evidenced by the successful execution of reactions in multi-liter setups described in the examples, demonstrating that the kinetics and heat transfer characteristics are manageable in standard stainless steel reactors, thereby facilitating a smooth transition to commercial tonnage production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of these imidazopyridine intermediates. The answers are derived directly from the experimental data and technical specifications provided in the patent literature, ensuring accuracy and relevance for industry professionals evaluating this technology for potential licensing or procurement.
Q: What is the primary therapeutic application of the imidazopyridine compounds described in this patent?
A: The compounds function as P2X3 receptor antagonists, showing significant potential for treating chronic cough, pain, and overactive bladder by inhibiting ATP-gated ion channels in sensory neurons.
Q: How does the patented process ensure regioselectivity during the acylation of polyfluorinated benzenes?
A: The process utilizes lithium diisopropylamide (LDA) at controlled low temperatures (-10 to 0°C) to generate a specific aryl lithium intermediate, which then reacts with the Weinreb amide to prevent over-addition and ensure precise substitution patterns.
Q: Is the synthetic route suitable for large-scale commercial manufacturing?
A: Yes, the method employs robust reactions such as palladium-catalyzed carbonylation and standard heterocyclic cyclization under mild conditions, avoiding extreme temperatures or hazardous reagents that typically hinder scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Imidazopyridine Compound Supplier
As the demand for P2X3 antagonists grows in the treatment of chronic cough and pain, securing a dependable source for high-quality intermediates is paramount. NINGBO INNO PHARMCHEM stands ready to support your development and commercialization goals with our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of this synthesis, including low-temperature lithiation and pressurized carbonylation, while adhering to stringent purity specifications. Our rigorous QC labs ensure that every batch of imidazopyridine intermediate meets the highest standards of quality, providing you with the confidence needed to advance your clinical programs.
We invite you to collaborate with us to leverage this innovative synthetic route for your supply chain. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this process can optimize your COGS. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us partner with you to bring these promising therapeutics to market efficiently and reliably.