Advanced Synthesis of Ceftaroline Side Chain Acid Trans Isomer for Commercial Scale-up
Advanced Synthesis of Ceftaroline Side Chain Acid Trans Isomer for Commercial Scale-up
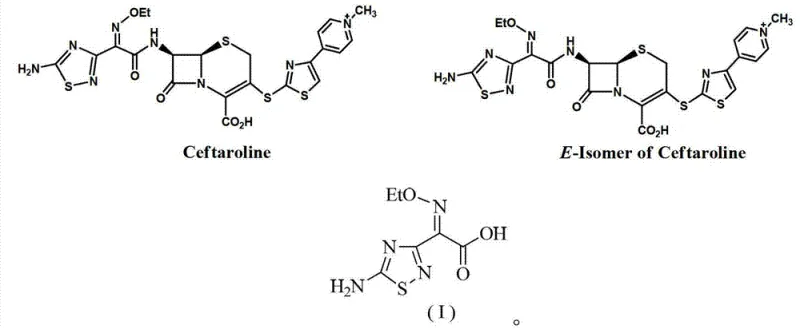
The pharmaceutical industry continuously seeks robust and efficient synthetic routes for critical antibiotic intermediates, particularly for fifth-generation cephalosporins like Ceftaroline. Patent CN103396380A introduces a groundbreaking preparation method for the trans-body of ceftaroline side chain acid, chemically known as (E)-2-(5-amino-1,2,4-thiadiazol-3-yl)-2-ethoxyiminoacetic acid. This specific intermediate is pivotal because the biological activity of cephalosporins is heavily dependent on the stereochemistry of the oxime side chain, where the (E)-isomer is the active form while the (Z)-isomer is often an inactive impurity. The disclosed technology addresses the long-standing challenge of obtaining high-purity trans-isomers without relying on inefficient separation techniques or unstable photochemical processes. By streamlining the synthesis into fewer steps with mild reaction conditions, this innovation offers a viable pathway for manufacturers aiming to enhance the purity profile of their final antibiotic products while optimizing production costs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of cephalosporin side chains containing oxime groups has been plagued by stereochemical inefficiencies. Traditional literature, such as reports from J. Antibiot. (1984), describes multi-step sequences involving cyclization, aminoacylation, oxidation, and carbonyl imidization, often resulting in the desired trans-isomer merely as a minor by-product with low overall yield. Another common approach involves synthesizing the Z-isomer first and attempting to convert it to the E-isomer via light irradiation. This photo-isomerization method is fundamentally flawed for large-scale operations due to the generation of numerous undefined by-products, difficult purification requirements, and unsatisfactory content levels of the target isomer. These legacy methods impose significant burdens on quality control laboratories and increase the cost of goods sold due to material loss during extensive purification cycles required to meet stringent pharmacopeial standards for impurity profiles.
The Novel Approach
In stark contrast to these cumbersome legacy protocols, the method disclosed in patent CN103396380A utilizes a direct and highly selective synthetic strategy. The core innovation lies in the direct oximation of a 2-(5-amino-1,2,4-thiadiazol-3-yl)acetamide derivative followed by a controlled etherification and hydrolysis sequence. This route bypasses the need for photochemical reactors and eliminates the reliance on separating difficult geometric isomers post-synthesis. The process begins with the reaction of the acetamide derivative with a nitrite ester under acidic catalysis, which inherently favors the formation of the thermodynamically stable (E)-oxime configuration. Subsequent etherification with an ethylating agent and final hydrolysis yields the target acid with exceptional purity. This streamlined approach not only reduces the number of unit operations but also significantly minimizes the formation of the unwanted cis-isomer, thereby simplifying downstream processing and enhancing the overall economic viability of the manufacturing process.

Mechanistic Insights into Oximation and Etherification Selectivity
The success of this synthetic route hinges on the precise control of stereoselectivity during the initial oximation step. When the 2-(5-amino-1,2,4-thiadiazol-3-yl)acetamide derivative reacts with a nitrite ester (such as isopropyl nitrite) in the presence of a hydrochloric acid catalyst, the reaction mechanism proceeds through a nitrosation intermediate that rapidly tautomerizes to the oxime. The specific electronic environment of the thiadiazole ring and the steric hindrance provided by the adjacent amide group favor the formation of the (E)-configuration over the (Z)-configuration. This kinetic and thermodynamic preference is crucial because it establishes the stereochemical integrity of the molecule early in the synthesis, preventing the accumulation of hard-to-remove geometric impurities. Furthermore, the use of specific organic solvents like tetrahydrofuran or ethyl acetate during this stage helps stabilize the transition state, ensuring that the reaction proceeds cleanly to completion without significant degradation of the sensitive heterocyclic core.
Following the oximation, the etherification step serves to protect the oxime hydroxyl group as an ethoxyimine, which is essential for the stability of the final cephalosporin antibiotic. This step involves the use of a base, such as triethylamine or potassium carbonate, to deprotonate the oxime, followed by nucleophilic attack on an ethylating agent like ethyl bromide or diethyl sulfate. The choice of base and solvent (e.g., acetonitrile or DMF) is critical to prevent O-to-N migration or hydrolysis of the amide protecting groups prematurely. Finally, the hydrolysis step employs an inorganic alkali, typically sodium hydroxide, to cleave the amide protecting groups and reveal the free amino group and carboxylic acid. The mild alkaline conditions (60°C to 90°C) are carefully selected to ensure complete deprotection without inducing racemization or degradation of the labile beta-lactam-like precursors, resulting in a final product with HPLC purity exceeding 98.0% and cis-isomer content below 0.5%.
How to Synthesize (E)-2-(5-amino-1,2,4-thiadiazol-3-yl)-2-ethoxyiminoacetic acid Efficiently
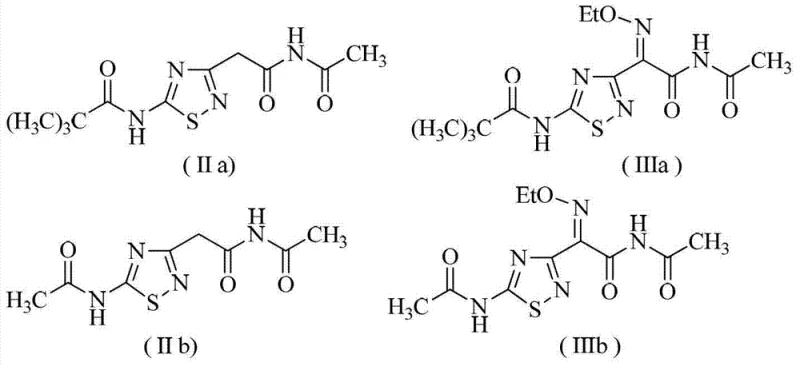
Implementing this synthesis requires careful attention to stoichiometry and temperature control to maximize yield and stereoselectivity. The process is designed to be operationally simple, utilizing standard chemical engineering equipment found in most fine chemical facilities. The initial oximation is conducted at moderate temperatures (25°C to 65°C) to balance reaction rate with selectivity, while the subsequent etherification is performed at slightly lower temperatures (5°C to 60°C) to minimize side reactions. The final hydrolysis step utilizes aqueous alkali solutions, which are cost-effective and easy to handle on a large scale. Detailed below is the standardized workflow derived from the patent examples, illustrating how specific derivatives (such as those with pivaloyl or acetyl protecting groups) are processed to yield the final high-purity acid. This protocol demonstrates the versatility of the method across different protecting group strategies, ensuring adaptability for various manufacturing setups.
- Oximation Reaction: React 2-(5-amino-1,2,4-thiadiazol-3-yl)acetamide derivative with nitrite ester in the presence of hydrochloric acid catalyst at 25°C to 65°C.
- Etherification Reaction: Treat the intermediate with an ethylating agent (e.g., ethyl bromide) and a base (e.g., triethylamine) in organic solvent at 5°C to 60°C.
- Hydrolysis: Hydrolyze the resulting amide derivative using an inorganic alkali aqueous solution at 30°C to 100°C to obtain the final acid product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route offers tangible strategic benefits beyond mere technical superiority. The primary advantage lies in the drastic simplification of the supply chain for raw materials. Unlike older methods that may require specialized photochemical equipment or exotic reagents, this process relies on commodity chemicals such as nitrite esters, ethyl bromide, and common inorganic bases. This shift to widely available feedstocks reduces the risk of supply disruptions and mitigates price volatility associated with niche reagents. Furthermore, the reduction in synthetic steps directly correlates to a reduction in solvent consumption and waste generation, aligning with modern green chemistry initiatives and reducing the environmental compliance burden on manufacturing sites. The ability to produce the intermediate with high stereoselectivity also means less material is wasted during purification, effectively increasing the throughput of existing production assets without the need for capital-intensive expansion.
- Cost Reduction in Manufacturing: The elimination of the photo-isomerization step removes the need for expensive UV reactors and the associated energy costs, while the higher yield per step reduces the overall cost of goods. By avoiding the generation of significant amounts of the inactive cis-isomer, the costly chromatographic separations or recrystallizations typically required to meet purity specifications are minimized or eliminated. This efficiency translates into substantial cost savings in both raw material consumption and waste disposal, allowing for more competitive pricing in the global API market.
- Enhanced Supply Chain Reliability: The reliance on stable, commercially available starting materials ensures a robust supply chain that is less susceptible to geopolitical or logistical shocks. The mild reaction conditions (mostly below 100°C) allow for production in standard glass-lined or stainless steel reactors, meaning that multiple contract manufacturing organizations (CMOs) can easily adopt this process without retrofitting their facilities. This flexibility increases the pool of potential suppliers, thereby reducing lead times and ensuring continuity of supply for critical antibiotic programs.
- Scalability and Environmental Compliance: The process generates fewer by-products and utilizes simpler work-up procedures, such as standard aqueous extractions and filtrations, which are easily scalable from pilot plant to multi-ton production. The reduced use of hazardous reagents and the avoidance of heavy metal catalysts simplify wastewater treatment and regulatory filings. This environmental friendliness not only lowers operational costs related to waste management but also future-proofs the manufacturing process against increasingly stringent global environmental regulations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the practical aspects of adopting this technology. Understanding these details is essential for R&D teams evaluating the feasibility of tech transfer and for quality assurance teams establishing specification limits for the new intermediate.
Q: How does this novel method improve stereoselectivity compared to traditional photo-isomerization?
A: Unlike traditional methods that rely on inefficient light irradiation of Z-isomers which produce many by-products, this patented route utilizes a direct oximation strategy that inherently favors the formation of the (E)-isomer, achieving HPLC content ≥98.0% with cis-isomer content <0.5%.
Q: What are the key raw materials required for this synthesis?
A: The process utilizes readily available starting materials including 2-(5-amino-1,2,4-thiadiazol-3-yl)acetamide derivatives, nitrite esters (such as isopropyl nitrite), and common ethylating agents like ethyl bromide or diethyl sulfate, ensuring stable supply chains.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the reaction conditions are mild (25°C to 90°C) and avoid hazardous reagents or extreme pressures. The simplified three-step sequence reduces operational complexity, making it highly scalable for commercial manufacturing of antibiotic intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ceftaroline Side Chain Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development and commercialization of life-saving antibiotics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering ceftaroline side chain acid and related pharmaceutical intermediates with stringent purity specifications, supported by our rigorous QC labs that utilize advanced analytical techniques to verify stereochemical integrity and impurity profiles. Our facility is equipped to handle the specific reaction conditions outlined in this patent, guaranteeing a consistent supply of material that meets the exacting standards of the global pharmaceutical industry.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic advantages of switching to this more efficient manufacturing process. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, allowing you to make informed decisions that enhance your supply chain resilience and product quality.