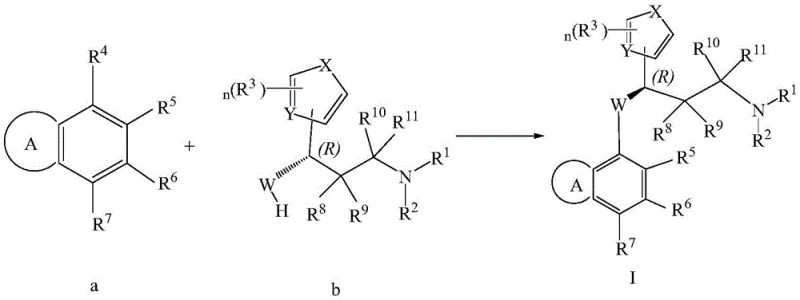
Scalable Synthesis of 3-Aryloxy-3-Heteroaryl-Propylamines for Next-Generation Pain Management Therapies
The pharmaceutical industry is constantly seeking robust and scalable methods to produce novel analgesic agents, particularly those targeting non-opioid pathways to mitigate the risks of addiction and tolerance. Patent CN115703767A introduces a significant advancement in the preparation of 3-aryloxy-3-five-membered heteroaryl-propylamine compounds, which serve as potent TRPA1 antagonists for the treatment of chronic and neuropathic pain. This technology addresses the critical need for efficient synthetic routes that can support the growing demand for non-addictive pain management solutions. By leveraging a streamlined etherification strategy followed by a precise deprotection sequence, this method offers a viable pathway for producing high-purity intermediates essential for modern drug development. The innovation lies not only in the chemical transformation but also in the operational simplicity that facilitates industrial adoption.
For research and development directors focusing on impurity profiles and process feasibility, the significance of this patent cannot be overstated. The described methodology circumvents the complexities often associated with constructing the chiral ether linkage found in these bioactive molecules. Traditional approaches frequently suffer from low yields or require harsh conditions that compromise the stereochemical integrity of the molecule. In contrast, the disclosed process utilizes mild alkaline conditions and specific catalytic systems to achieve high conversion rates while maintaining the crucial (R)-configuration. This level of control is paramount for ensuring the biological efficacy of the final therapeutic agent, as TRPA1 receptors are highly sensitive to stereoisomerism. The ability to consistently produce the correct enantiomer reduces the burden on downstream purification and ensures a cleaner impurity profile.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex aryl ether derivatives has been plagued by significant technical hurdles that impact both cost and timeline. Conventional methods often rely on expensive transition metal catalysts or require extreme temperatures that can lead to decomposition of sensitive functional groups. Furthermore, many existing routes involve multiple protection and deprotection steps that add unnecessary length to the synthetic sequence, thereby increasing the accumulation of byproducts and reducing overall throughput. These inefficiencies create bottlenecks in the supply chain, making it difficult to secure reliable quantities of high-quality intermediates for clinical trials. The reliance on hazardous reagents also poses environmental and safety challenges that complicate regulatory approval and waste management protocols. Consequently, manufacturers face elevated production costs and extended lead times, which ultimately delay the availability of life-saving medications to patients in need of effective pain relief.
The Novel Approach
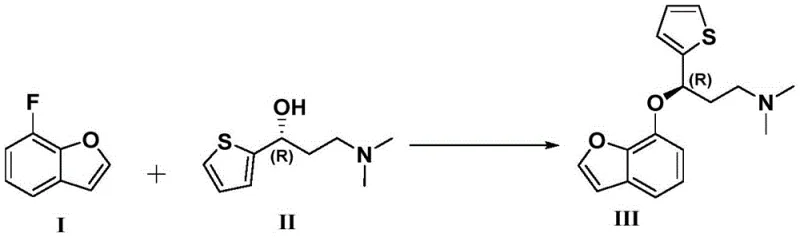
The methodology outlined in CN115703767A represents a paradigm shift towards greener and more efficient chemical manufacturing. By employing potassium iodide as a catalyst in dimethyl sulfoxide, the reaction proceeds smoothly at moderate temperatures ranging from 55°C to 65°C. This eliminates the need for energy-intensive heating or cryogenic cooling, substantially lowering the operational footprint. The direct nucleophilic substitution between the fluorobenzofuran derivative and the chiral amino-alcohol is highly selective, minimizing the formation of regioisomers. Additionally, the subsequent N-demethylation strategy using phenyl chloroformate provides a controlled means to modify the amine functionality without affecting the sensitive ether bond. This two-stage refinement ensures that the final product meets stringent purity specifications required for pharmaceutical applications. The overall process design prioritizes atom economy and safety, aligning with modern principles of sustainable chemistry.
Mechanistic Insights into KI-Catalyzed Nucleophilic Substitution
At the heart of this synthesis is a finely tuned nucleophilic aromatic substitution mechanism that leverages the activating effect of the heteroaryl ring. The presence of the fluorine atom on the benzofuran ring serves as an excellent leaving group when activated by the electron-withdrawing nature of the adjacent oxygen atom. The addition of potassium iodide enhances the nucleophilicity of the alcohol oxygen through halogen exchange interactions, facilitating the attack on the aromatic carbon. This catalytic cycle is crucial for driving the reaction to completion without requiring stoichiometric amounts of expensive reagents. The reaction environment is carefully buffered with sodium hydroxide to deprotonate the alcohol, generating the reactive alkoxide species in situ. This precise control over the basicity prevents side reactions such as elimination or hydrolysis of the nitrile or ester groups that might be present on the scaffold. Understanding this mechanistic nuance allows chemists to optimize reaction parameters for maximum efficiency.
Furthermore, the preservation of chirality during this transformation is a critical aspect of the mechanism. The reaction conditions are sufficiently mild to prevent racemization at the benzylic position, which is susceptible to epimerization under strong acidic or basic conditions. The use of polar aprotic solvents like DMSO stabilizes the transition state and solvates the cationic species, allowing the anionic nucleophile to react freely. Following the initial coupling, the intermediate undergoes a protection step where the secondary amine is converted into a carbamate. This temporary modification shields the nitrogen from unwanted oxidation or alkylation during subsequent processing. The final hydrolysis step cleaves this protecting group under controlled alkaline conditions, regenerating the free amine while leaving the ether linkage intact. This sequential logic ensures that each functional group is manipulated with high specificity, resulting in a final product with a well-defined structural identity.
How to Synthesize 3-Aryloxy-3-Heteroaryl-Propylamine Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires strict adherence to the specified reaction parameters to ensure reproducibility and safety. The process begins with the dissolution of the fluorinated starting material in dry DMSO, followed by the sequential addition of the chiral alcohol, catalyst, and base. Temperature control is vital during the exothermic addition of the base to prevent thermal runaway. Once the etherification is complete, the reaction mixture is quenched and extracted using ethyl acetate, a solvent chosen for its favorable partition coefficients and ease of removal. The resulting crude intermediate is then subjected to the protection step in dichloromethane, where temperature is maintained below 30°C to avoid decomposition of the chloroformate reagent. Finally, the hydrolysis is performed in a mixed solvent system to ensure homogeneity while driving the cleavage of the carbamate bond. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in replicating these results.
- Perform nucleophilic substitution of 7-fluorobenzofuran with chiral thiophene-propanol using KI catalyst in DMSO.
- Execute N-demethylation protection via reaction with phenyl chloroformate in dichloromethane.
- Complete hydrolysis of the carbamate intermediate using sodium hydroxide to yield the final amine product.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this synthetic route offers substantial benefits by utilizing commodity chemicals that are readily available in the global market. The primary reagents, such as 7-fluorobenzofuran and potassium iodide, are produced by multiple suppliers, reducing the risk of single-source dependency. This diversification of the supply base enhances resilience against market fluctuations and logistical disruptions. Moreover, the elimination of precious metal catalysts removes a significant cost driver and simplifies the purification process, as there is no need for specialized scavenging resins to remove trace metals. The use of common solvents like DMSO and ethyl acetate further streamlines inventory management and waste disposal, as these materials are handled routinely in most chemical facilities. These factors collectively contribute to a more predictable and stable cost structure for the manufacturing of these valuable intermediates.
- Cost Reduction in Manufacturing: The process achieves cost efficiency through the use of inexpensive inorganic bases and catalysts rather than costly organometallic complexes. By operating at atmospheric pressure and moderate temperatures, energy consumption is significantly minimized compared to high-pressure hydrogenation or cryogenic reactions. The high conversion rates reported in the patent examples indicate that raw material utilization is optimized, reducing the volume of waste generated per kilogram of product. This efficiency translates directly into lower variable costs per unit, allowing for more competitive pricing strategies in the B2B marketplace. Additionally, the simplified workup procedure reduces labor hours and equipment occupancy time, further enhancing the overall economic viability of the production campaign.
- Enhanced Supply Chain Reliability: The reliance on stable, shelf-stable reagents ensures that production schedules can be maintained without interruption due to reagent degradation. The robustness of the reaction conditions means that the process is less sensitive to minor variations in raw material quality, providing a buffer against supply chain inconsistencies. This reliability is crucial for meeting the strict delivery timelines required by pharmaceutical clients who operate on tight development schedules. By establishing a manufacturing process that is resilient to external variables, suppliers can offer greater assurance of continuity of supply. This stability fosters stronger long-term partnerships with clients who prioritize dependability alongside technical performance in their vendor selection criteria.
- Scalability and Environmental Compliance: The synthetic pathway is inherently scalable, having been demonstrated effectively from gram to kilogram scales in the patent examples. The absence of hazardous gases or highly unstable intermediates simplifies the engineering controls required for scale-up, reducing capital expenditure on specialized containment systems. Furthermore, the aqueous workup and extraction steps generate waste streams that are easier to treat and dispose of in compliance with environmental regulations. The reduced use of chlorinated solvents in the final steps aligns with green chemistry initiatives, improving the sustainability profile of the manufacturing site. This alignment with environmental standards mitigates regulatory risk and enhances the corporate social responsibility standing of the manufacturing organization.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented technology. They are derived from the specific experimental data and beneficial effects described in the patent documentation. Understanding these details helps stakeholders evaluate the feasibility of integrating this chemistry into their existing pipelines. The answers reflect the objective capabilities of the method as disclosed, providing a clear basis for decision-making.
Q: What are the key advantages of this synthesis method over conventional routes?
A: This method utilizes mild reaction conditions (55-65°C) and inexpensive catalysts like potassium iodide, significantly simplifying the operational complexity compared to traditional high-temperature or heavy-metal catalyzed processes.
Q: How is chiral integrity maintained during the etherification step?
A: The process employs a specific chiral starting material ((R)-3-(dimethylamino)-1-(thiophen-2-yl)propan-1-ol) and mild basic conditions that prevent racemization, ensuring the final product retains the required stereochemistry for TRPA1 binding.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the use of common solvents like DMSO and ethyl acetate, along with ambient pressure reactions and straightforward workup procedures involving acid-base extraction, makes this route highly adaptable for multi-kilogram to ton-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Aryloxy-3-Heteroaryl-Propylamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of securing a stable supply of high-quality pharmaceutical intermediates for the development of next-generation analgesics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from clinical trials to market launch. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch meets the highest international standards. Our facility is equipped to handle the specific solvent systems and reaction conditions required for this synthesis, providing a safe and compliant environment for manufacturing. By partnering with us, you gain access to a supply chain that is both technically proficient and commercially responsive to your evolving needs.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can benefit your specific project requirements. We are prepared to provide a Customized Cost-Saving Analysis that evaluates the economic impact of adopting this method for your production volumes. Please contact us to request specific COA data and route feasibility assessments tailored to your target molecules. Our goal is to support your R&D efforts with reliable materials and expert insights, enabling you to bring effective pain management therapies to patients faster and more efficiently. Let us collaborate to advance the future of non-opioid pain treatment through superior chemical manufacturing.