Advanced Synthesis of Naphthoyl Amine Derivatives for Kinase Inhibitor Manufacturing
Advanced Synthesis of Naphthoyl Amine Derivatives for Kinase Inhibitor Manufacturing
The pharmaceutical landscape is increasingly demanding versatile scaffolds capable of dual-target inhibition, particularly for complex oncology and metabolic disorders. Patent CN102260210A introduces a groundbreaking preparation method for naphthoyl amine derivatives, specifically designed as potent protein kinase and histone deacetylase (HDAC) inhibitors. These compounds, represented by Formula I, exhibit a unique structural architecture combining a substituted quinoline moiety with a naphthalene core via an ether linkage. This specific arrangement is critical for binding affinity and biological activity against a broad spectrum of diseases, including cardiovascular disorders, cancers, and hormone-related conditions. As a leading entity in fine chemical manufacturing, we recognize the immense potential of this scaffold in next-generation drug discovery pipelines. The patent details a robust, three-step synthetic strategy that bypasses the limitations of earlier methodologies, offering a clear path toward high-purity intermediates essential for clinical development.
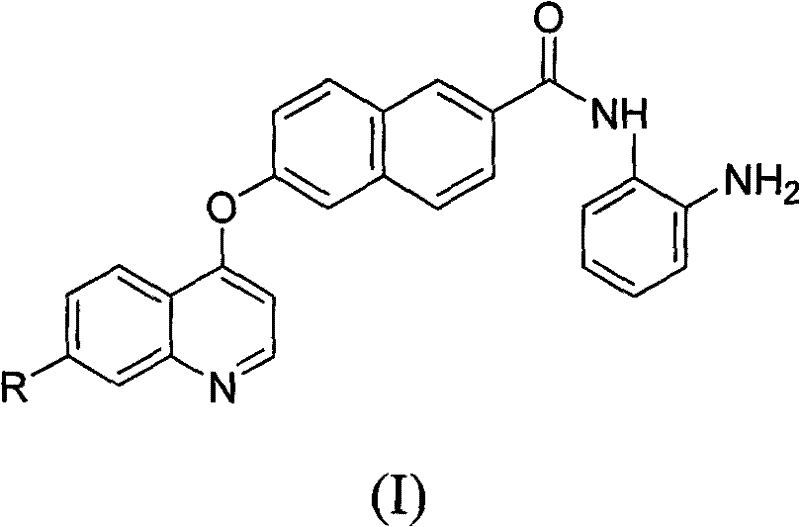
For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediate supplier, understanding the nuances of this synthesis is paramount. The disclosed method not only ensures structural integrity but also optimizes the process for scalability. By leveraging accessible starting materials such as 6-hydroxy-2-naphthoic acid and various 4-chloroquinoline derivatives, the process minimizes supply chain risks associated with exotic reagents. Furthermore, the reaction conditions are meticulously defined to maximize yield while maintaining safety standards, making this technology a cornerstone for cost reduction in API manufacturing. The ability to introduce diverse substituents (R=F, Cl, Br, I, OCH3, etc.) at the 7-position of the quinoline ring allows for extensive Structure-Activity Relationship (SAR) studies without overhauling the core synthetic route.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex heterocyclic amides involving naphthalene and quinoline systems has been plagued by inefficiencies. Traditional routes often rely on multi-step sequences involving protection and deprotection strategies, which drastically increase material costs and waste generation. Many conventional methods utilize expensive transition metal catalysts for C-O or C-N bond formation, necessitating rigorous purification steps to remove trace metal impurities that are unacceptable in pharmaceutical grades. Additionally, older protocols frequently suffer from poor regioselectivity, leading to difficult-to-separate isomers that compromise the overall purity of the final active pharmaceutical ingredient (API). The reliance on harsh conditions or unstable intermediates in legacy processes often results in low throughput, creating bottlenecks in the supply chain for high-purity kinase inhibitor intermediates. These factors collectively inflate the cost of goods sold (COGS) and extend the lead time for drug candidates entering clinical trials.
The Novel Approach
The methodology outlined in CN102260210A represents a paradigm shift towards streamlined efficiency. By employing a direct nucleophilic aromatic substitution followed by activation and amidation, the process eliminates unnecessary synthetic steps. The use of sodium hydride (NaH) in dimethylformamide (DMF) facilitates a clean etherification between the naphthoic acid and the chloroquinoline derivative, establishing the core backbone in a single pot. This is followed by a straightforward conversion to an acyl chloride using thionyl chloride, a reagent known for its effectiveness and ease of removal. The final coupling with o-phenylenediamine proceeds under mild conditions in tetrahydrofuran (THF), ensuring the stability of sensitive functional groups. This novel approach significantly simplifies the operational complexity, allowing for the commercial scale-up of complex heterocyclic compounds with minimal equipment modification. The result is a process that is not only chemically elegant but also economically superior, addressing the critical need for reducing lead time for high-purity pharmaceutical intermediates.
Mechanistic Insights into the Three-Step Synthetic Route
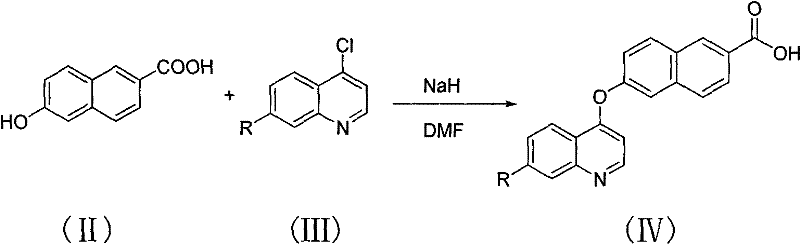
The first critical transformation involves the formation of the ether linkage, which serves as the structural bridge between the naphthalene and quinoline systems. In this step, 6-hydroxy-2-naphthoic acid is deprotonated by sodium hydride (2.5 equivalents) in anhydrous DMF under an ice bath to prevent exothermic runaway. Once the phenoxide anion is generated, the temperature is allowed to return to ambient before heating to 110°C for 6-10 hours in the presence of the 4-chloroquinoline derivative. This thermal energy drives the nucleophilic attack on the electron-deficient carbon of the quinoline ring, displacing the chloride ion. The reaction mixture is then quenched in ice water and carefully adjusted to pH 6.5 with hydrochloric acid to precipitate the intermediate carboxylic acid (Formula IV) as a yellow solid. This precise pH control is vital for maximizing recovery and minimizing the loss of product in the aqueous phase.
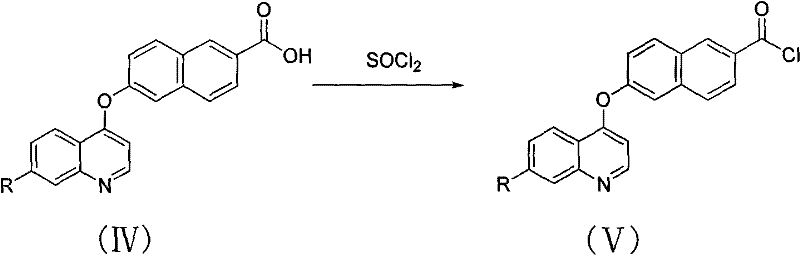
Following the isolation of the carboxylic acid intermediate, the second step focuses on activating the carbonyl group for subsequent amidation. The dried Formula IV compound is suspended in excess thionyl chloride (SOCl2) and subjected to reflux for 5-7 hours. This prolonged heating ensures the complete conversion of the carboxylic acid to the corresponding acid chloride (Formula V). The mechanism involves the nucleophilic attack of the carbonyl oxygen on the sulfur atom of SOCl2, followed by the elimination of SO2 and HCl gases. Upon completion, the excess thionyl chloride is removed via distillation under reduced pressure, yielding the reactive acid chloride without the need for further purification. This in-situ generation of the electrophile is a strategic move to prevent hydrolysis and maintain high reactivity for the final coupling step.
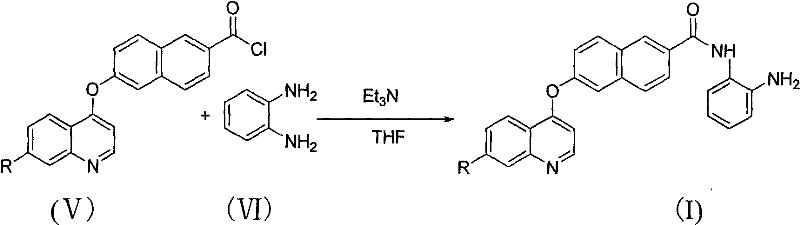
The final stage of the synthesis is the formation of the amide bond, which installs the aniline moiety essential for biological activity. The acid chloride (Formula V) is dissolved in THF and reacted with o-phenylenediamine (1.0 equivalent) in the presence of triethylamine (2.0 equivalents) as a proton scavenger. The reaction is initiated at 0°C to control the exotherm and then allowed to proceed at 25-50°C for 6-8 hours. Triethylamine neutralizes the HCl generated during the nucleophilic attack of the amine on the acid chloride, driving the equilibrium toward product formation. Post-reaction workup involves filtration to remove triethylamine hydrochloride salts, followed by extraction with ethyl acetate and drying over anhydrous sodium sulfate. The crude product is finally recrystallized from methylene chloride to afford the target Formula I compound with high purity, ready for biological evaluation.
How to Synthesize Naphthoyl Amine Derivatives Efficiently
Executing this synthesis requires strict adherence to the specified stoichiometry and temperature profiles to ensure reproducibility and safety. The process is designed to be robust, tolerating a variety of substituents at the R-position without significant deviation in protocol. Operators must ensure that all solvents are anhydrous, particularly for the NaH and SOCl2 steps, to prevent side reactions that could lower yield. The detailed standardized operating procedures for each stage, including specific quenching methods and crystallization parameters, are critical for maintaining batch-to-batch consistency. For a comprehensive guide on the exact experimental execution, please refer to the technical breakdown below.
- Perform nucleophilic aromatic substitution between 6-hydroxy-2-naphthoic acid and 4-chloroquinoline derivatives using NaH in DMF at 110°C.
- Convert the resulting carboxylic acid intermediate to an acyl chloride using thionyl chloride (SOCl2) under reflux conditions.
- React the acyl chloride with o-phenylenediamine in THF using triethylamine as a base to form the final naphthoyl amine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers substantial advantages that directly impact the bottom line and supply chain resilience. The elimination of precious metal catalysts removes a significant cost center and simplifies the regulatory burden associated with heavy metal clearance in final drug products. By utilizing commodity chemicals like thionyl chloride and triethylamine, the process leverages established global supply chains, ensuring consistent availability of raw materials even during market fluctuations. The simplicity of the workup procedures, primarily involving precipitation and filtration rather than complex chromatography, reduces solvent consumption and waste disposal costs. This efficiency translates into a more sustainable manufacturing profile, aligning with modern environmental, social, and governance (ESG) goals.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by avoiding expensive catalytic systems and reducing the total number of unit operations. The use of inexpensive reagents like sodium hydride and thionyl chloride, combined with high-yielding steps, lowers the overall cost of goods. Furthermore, the ability to recycle solvents like DMF and THF through standard distillation processes contributes to long-term operational savings. The streamlined nature of the synthesis reduces labor hours and equipment occupancy time, enhancing overall plant throughput and economic viability.
- Enhanced Supply Chain Reliability: Sourcing risk is minimized as all key starting materials, including 6-hydroxy-2-naphthoic acid and substituted chloroquinolines, are commercially available from multiple vendors. The robustness of the reaction conditions means that minor variations in raw material quality can be accommodated without compromising the final product specification. This flexibility ensures a continuous supply of intermediates, preventing production delays that could impact downstream API manufacturing schedules. The scalability of the process allows for rapid ramp-up from kilogram to tonne scales to meet fluctuating market demands.
- Scalability and Environmental Compliance: The synthetic pathway is inherently scalable, with no steps requiring specialized high-pressure or cryogenic equipment beyond standard industrial capabilities. The generation of byproducts is manageable, with gaseous emissions like SO2 and HCl easily scrubbed using standard abatement systems. The solid waste generated is primarily inorganic salts, which can be disposed of or treated according to standard industrial protocols. This compliance-friendly profile facilitates faster regulatory approvals and reduces the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of these naphthoyl amine derivatives. Understanding these details helps stakeholders make informed decisions about integrating this technology into their development pipelines. The answers are derived directly from the patented methodology and practical manufacturing experience.
Q: What are the critical reaction conditions for the etherification step?
A: The etherification requires strict temperature control, initiating at 0°C with NaH addition, warming to room temperature, and finally heating to 110°C for 6-10 hours in DMF to ensure complete conversion.
Q: How is purity maintained during the final amidation step?
A: Purity is ensured by using stoichiometric amounts of o-phenylenediamine and triethylamine in THF, followed by rigorous workup including filtration, extraction, and recrystallization from methylene chloride.
Q: Is this synthesis route suitable for large-scale production?
A: Yes, the process utilizes readily available raw materials and standard solvents like DMF and THF, avoiding expensive transition metal catalysts, which makes it highly suitable for industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Naphthoyl Amine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we possess the technical expertise and infrastructure to bring this sophisticated chemistry from the laboratory to commercial reality. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. We operate stringent purity specifications and maintain rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee the quality of every batch. Our commitment to excellence extends beyond mere synthesis; we offer comprehensive support in process optimization and regulatory documentation to facilitate your drug approval journey.
We invite you to collaborate with us to leverage this advanced synthetic route for your oncology or metabolic disease programs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can accelerate your project timelines and reduce overall development costs.