Advanced Synthesis of Indole-Propionic Acid Derivatives for Pharmaceutical Applications
Advanced Synthesis of Indole-Propionic Acid Derivatives for Pharmaceutical Applications
The pharmaceutical industry is constantly seeking more efficient and sustainable pathways to access critical building blocks for novel therapeutics, particularly in the realm of immuno-oncology. Patent CN111303001B, published in late 2022, introduces a groundbreaking synthesis method for indole mono-substituted 2-hydroxy-3-(1H-indol-3-yl) propionic acid compounds. These molecules serve as pivotal intermediates in the construction of IDO (Indoleamine 2,3-dioxygenase) and TDO (Tryptophan 2,3-dioxygenase) inhibitors, which are gaining immense traction as cancer immunotherapy agents. The disclosed technology represents a significant departure from traditional biocatalytic or harsh chemical methods, offering a streamlined, two-step chemical protocol that utilizes inexpensive, commercially available starting materials. By leveraging a Friedel-Crafts-type alkylation followed by a diazotization-hydrolysis sequence, this innovation addresses the critical bottlenecks of cost, safety, and scalability that have long plagued the supply chain for these high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of tryptophan derivatives and related indole-propionic acids has relied on methodologies that are either economically prohibitive or environmentally unsustainable. One prominent prior art route, detailed in literature such as the Journal of the American Chemical Society (2018), involves the reaction of indole with methyloxirane-2-carboxylic acid esters. As illustrated in the reaction scheme below, this pathway necessitates the use of stoichiometric amounts of tin tetrachloride (SnCl4) and carbon tetrachloride (CCl4), reagents that are not only expensive but also pose severe toxicity and disposal challenges.
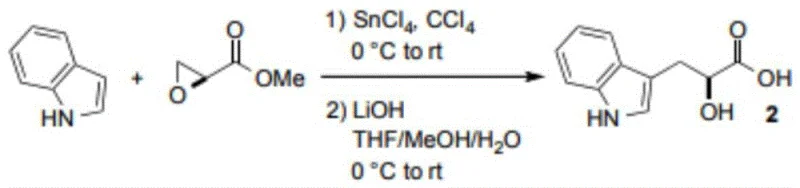
Furthermore, alternative biological enzyme methods, while selective, suffer from high production costs due to the expense of enzyme preparation and the complexity of maintaining sterile, controlled fermentation environments. Chemical routes involving hippuric acid condensation or giant reed alkali reactions often require harsh conditions, multiple protection-deprotection steps, and result in poor atom economy. These legacy methods create substantial barriers for procurement teams aiming to secure reliable supplies of high-purity intermediates at a viable cost point for commercial drug manufacturing.
The Novel Approach
In stark contrast, the method disclosed in CN111303001B simplifies the synthetic landscape by employing a direct condensation strategy. The novel approach utilizes substituted indoles (Formula I) and DL-serine as the primary feedstocks, reacting them in the presence of acetic anhydride and a carboxylic acid solvent. This eliminates the need for exotic epoxides and toxic Lewis acids entirely. The process is operationally simple, proceeding under inert gas protection with standard heating, making it inherently safer and more compatible with existing chemical infrastructure. By shifting the paradigm from complex multi-step sequences to a concise two-step transformation, this technology offers a robust solution for cost reduction in API manufacturing, ensuring a more stable and predictable supply chain for downstream drug developers.
Mechanistic Insights into Serine-Mediated Indole Alkylation and Diazotization
The core of this technological breakthrough lies in the clever activation of DL-serine to function as an electrophile for the indole nucleus. In the first step, acetic anhydride reacts with the hydroxyl group of serine, generating a reactive acetate species in situ. This activated intermediate undergoes an electrophilic aromatic substitution with the electron-rich indole ring at the C3 position. The reaction is facilitated by the acidic medium (glacial acetic acid), which protonates the leaving group and stabilizes the transition state. Following the alkylation, a subsequent alkaline heat treatment hydrolyzes the acetyl protecting groups and facilitates the formation of the stable amino-acid intermediate (Formula II). This mechanism ensures high regioselectivity for the 3-position of the indole, minimizing the formation of unwanted 2-substituted isomers that are difficult to separate.
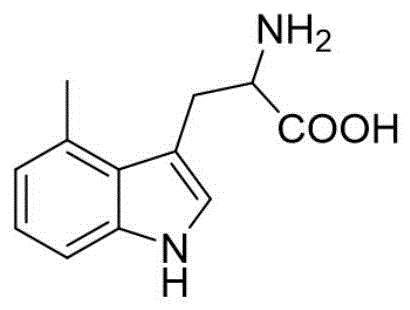
The second mechanistic phase involves the conversion of the primary amine into a hydroxyl group via a diazonium salt. Under strongly acidic conditions provided by sulfuric acid and low temperatures (-5 to 0°C), sodium nitrite generates nitrous acid, which diazotizes the amino group of the intermediate. This unstable diazonium species is then subjected to controlled heating (65-75°C) in an aqueous environment. The thermal energy promotes the heterolytic cleavage of the C-N bond, releasing nitrogen gas and allowing a water molecule to attack the carbocation, thereby installing the hydroxyl functionality. This classic Sandmeyer-type hydrolysis is executed with remarkable efficiency in this patent, avoiding the radical side reactions that often plague such transformations in complex heterocyclic systems.
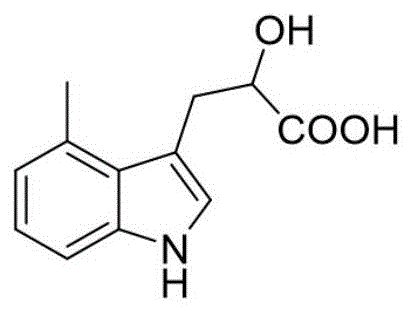
From an impurity control perspective, the choice of mild acidic conditions in Step 1 and the precise temperature control in Step 2 are critical. The use of glacial acetic acid prevents the polymerization of the indole ring, a common side reaction under stronger mineral acid conditions. Furthermore, the workup procedures involving pH adjustment and solvent extraction are specifically tuned to separate the zwitterionic product from unreacted starting materials and inorganic salts. This rigorous control over the reaction environment ensures that the final product meets the stringent purity specifications required for clinical-grade pharmaceutical intermediates, reducing the burden on downstream purification processes.
How to Synthesize 2-hydroxy-3-(1H-indol-3-yl) propionic acid Efficiently
The practical implementation of this synthesis route is designed for ease of execution in standard chemical reactors. The process begins with the dissolution of the substituted indole and DL-serine in glacial acetic acid, followed by the slow addition of acetic anhydride. The mixture is refluxed for approximately 10 to 14 hours, allowing the alkylation to reach completion. After removing the solvent, the residue is treated with an aqueous alkali solution to effect hydrolysis. The resulting amino-acid intermediate is isolated via extraction and chromatography. In the second stage, this intermediate is dissolved in dilute sulfuric acid and cooled in an ice bath before the addition of aqueous sodium nitrite. After stirring to ensure complete diazotization, the mixture is heated to facilitate hydrolysis, yielding the target hydroxy acid after a final extraction and purification step.
- React substituted indole with DL-serine and acetic anhydride in glacial acetic acid at 65-85°C to form the amino-acid intermediate.
- Perform diazotization on the amino-acid intermediate using sodium nitrite and sulfuric acid at low temperature (-5 to 0°C).
- Hydrolyze the diazonium salt by heating to 65-75°C to yield the final 2-hydroxy-3-(1H-indol-3-yl) propionic acid derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis method translates into tangible strategic benefits beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the raw material portfolio. By replacing scarce and regulated reagents like tin tetrachloride and carbon tetrachloride with commodity chemicals like acetic acid and DL-serine, the supply chain becomes significantly more resilient. These commodity feedstocks are produced globally at massive scales, insulating the manufacturing process from the volatility and geopolitical risks associated with specialty reagent sourcing. This shift ensures a continuous and reliable flow of materials, which is essential for maintaining uninterrupted production schedules in the fast-paced pharmaceutical sector.
- Cost Reduction in Manufacturing: The elimination of expensive Lewis acid catalysts and toxic solvents directly impacts the bill of materials. Tin salts and chlorinated solvents carry high acquisition costs and even higher disposal fees due to their classification as hazardous waste. By utilizing a benign acetic acid system, the process significantly reduces the operational expenditure related to waste management and environmental compliance. Furthermore, the high yields reported in the patent examples (consistently ranging from 76% to 85%) mean that less raw material is wasted per kilogram of product, driving down the effective cost per unit and enhancing the overall economic viability of the drug candidate.
- Enhanced Supply Chain Reliability: The reliance on DL-serine, a bulk fermentation product, and widely available substituted indoles ensures that the supply chain is not dependent on single-source suppliers of niche chemicals. This diversification of supply sources mitigates the risk of shortages that can delay clinical trials or commercial launches. Additionally, the robustness of the reaction conditions—tolerating standard heating and stirring without the need for cryogenic temperatures or ultra-high pressures—means that the process can be transferred easily between different manufacturing sites or CDMO partners without extensive re-validation, further securing the supply continuity.
- Scalability and Environmental Compliance: As the demand for IDO inhibitors grows, the ability to scale production from grams to tons is paramount. This synthetic route is inherently scalable because it avoids exothermic runaways associated with strong Lewis acids and uses solvents that are easy to recover and recycle. The aqueous workup steps simplify the isolation of the product, reducing the volume of organic solvents required for purification. From an environmental standpoint, the reduction in heavy metal usage aligns perfectly with the green chemistry initiatives mandated by major regulatory bodies, facilitating smoother regulatory approvals and enhancing the corporate sustainability profile of the manufacturing partner.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms legacy routes in terms of safety, efficiency, and product quality. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this intermediate into their broader drug development pipelines.
Q: Why is this new synthesis method superior to the JACS 2018 route?
A: The conventional route described in JACS 2018 utilizes toxic and expensive reagents like SnCl4 and CCl4, creating significant environmental and disposal costs. The new method replaces these with benign acetic acid and standard mineral acids, drastically reducing hazardous waste generation and raw material costs.
Q: What is the purity profile of the intermediates generated?
A: The process utilizes mild reaction conditions (65-85°C) and standard purification techniques like silica gel chromatography, which effectively remove side products. The patent demonstrates consistent yields between 76% and 85%, indicating a clean reaction profile suitable for pharmaceutical grade requirements.
Q: Can this method be scaled for industrial production of IDO inhibitors?
A: Yes, the method relies on readily available commodity chemicals like DL-serine and substituted indoles. The absence of sensitive organometallic catalysts and the use of standard aqueous workups make the process highly amenable to multi-kilogram and ton-scale manufacturing without specialized equipment.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Indole-Propionic Acid Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of novel oncology therapies depends on the availability of high-quality, cost-effective intermediates. Our team of expert chemists has thoroughly analyzed the methodology disclosed in CN111303001B and is fully prepared to execute this advanced synthesis at scale. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition seamlessly from clinical supply to full-scale manufacturing. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of indole-propionic acid derivative we deliver supports the efficacy and safety of your final drug product.
We invite you to collaborate with us to leverage this innovative synthetic route for your next-generation immunotherapy programs. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our manufacturing capabilities can accelerate your timeline to market while optimizing your overall production budget.